Introduction
Long-term results after orthopedic or surgical treatment of hemifacial microsomia (HFM) have shown a tendency toward recurrence of the facial asymmetry. However, the literature contains a number of successful case reports that show surprising changes in the morphology of the condyles. In addition, patients with similar mandibular asymmetries, treated early with surgery, have excellent long-term follow-ups, especially those who have little or no soft-tissue involvement, but only severe mandibular ramal deformities. The phenotypes of these cases are unexpectedly similar, with a consistent collapse of the condyle against the coronoid and a deep sigmoid notch. The objectives of this article were to help distinguish true HFM from this peculiar type of hemimandibular asymmetry morphologically and to quantify their differences before treatement and in the long term.
Methods
Panoramic radiographs taken at pretreatment and the long-term follow-up of 9 patients with hemimandibular hypoplasia, characterized by the collapse of the condyle against the coronoid, were compared with those of 8 patients with severe type I and type II HFM; these records were collected before and at least 10 years after distraction osteogenesis.
Results
Ratios and angular measurements before and after treatment differed significantly between the 2 groups.
Conclusions
Perhaps these patients were misdiagnosed and actually had secondary injuries of the condyle, which have a normal functional matrix. Therefore, with growth and functional stimulation, they would tend to grow toward the original symmetry. To make a differential diagnosis between true HFM and this peculiar type of hemimandibular hypoplasia, the collaboration between not only orthodontists and surgeons, but also geneticists and dysmorphologists, is of great importance because of the different prognoses.
Etiologic diagnosis is possibly the most difficult, but also the most important, step in orthodontic treatment. Facial asymmetries are certainly a challenging chapter for both the orthodontist and the maxillofacial surgeon. Hemifacial microsomia (HFM), the best known of the branchial arch syndromes, is a relatively common craniofacial anomaly with a birth prevalence of at least 1 in 5600, characterized by asymmetric underdevelopment of the structures originating from the first and second branchial arches. Deformities can involve the ear, the mandible, the maxilla, the zygomatic arch, the temporal bone, the fifth and eighth cranial nerves, the cervical spine, and the facial muscles. The degree of ear involvement is markedly variable. Ear tags and pits might be present. The condition is etiologically heterogeneous. Many chromosome abnormalities have been recorded, but also environmental causes including thalidomide, primidione, and retinoic acid administered during the organogenesis. A recent model, based on a mutation of a locus on chromosome 10, appears to support the hypothesis that HFM anomalies have partly a genetic cause. Most cases are sporadic, but rare familial instances with autosomal dominant inheritance have also been observed. HFM is 1 of 4 conditions defined as otofacial malformations, which are neurocristopathies, sharing a major involvement of neural crest cells, together with DiGeorge syndrome, retinoic acid syndrome, and Treacher Collins syndrome. A condition analogous to HFM has been induced in mice by causing a local hemorrhage from the embryonic stapedial artery between the 30th and 40th days of fetal develop-ment, a critical period of neural crest cell migration. Neural crest cells are a migratory cell population. Just before the neural fold fuses to form the neural tube, neuroectodermal cells adjacent to the neural plate migrate into the facial region, where they form the skeletal and connective tissues of the face: bone cartilage, fibrous connective tissue, and all dental tissues except enamel. Thus, facial mesenchyme is of neural crest origin; this in turn guides the formation of vascular endothelium and skeletal muscles, which are of mesodermal origin. Thus, a craniofacial malformation can be the consequence of a disruption in the migration or proliferation of neural crest cells, but the primary defect might have a genetic origin.
What is more important than the etiology, in view of the aim of this article, is that the neural crest cells that migrate into the first branchial arch carry the pattern of information needed for proper morphogenesis of mesodermal derivates such as cranial muscles. Extirpation of the mandibular neural crest stream leads to severe alterations of mandibular-muscle patterning. Whereas Meckel’s, palatoquadrate, suprarostral, and infrarostral cartilages can be severely malformed or missing, all muscles of the levator mandibulae group will also be similarly affected. Therefore, although HFM can be variable in terms of phenotype, mandibular deformity is always proportionate to the associated muscular deformity.
As stated before, under the diagnosis of HFM, there is much variability; thus, treatment varies. Patients with HFM with minor mandibular and soft-tissue involvement can be treated by using a conservative approach, obtaining good dentoalveolar correction and some mild improvement in the skeletal asymmetry. Orthodontic functional appliance therapy during growth has been suggested even in moderate to severe cases, but there is no consensus about the value and the true effect of such treatment.
At present, the timing of treatment and the optimal treatment protocol are still controversial. Therapy includes orthodontic and surgical measures for the correction of the skeletal asymmetry. Preoperative and postoperative treatment with functional appliances has been recommended to improve muscle function and to stimulate growth of the soft and hard tissues. Vargervik et al demonstrated that functional appliances improved the short-term results after costochondral grafting, but, nevertheless, in most cases, the asymmetry returned during subsequent growth. Likewise, it was recently shown that functional therapy associated with distraction osteogenesis only slows down the return to the original asymmetry of HFM. In contrast, the literature includes many case reports describing successful orthopedic treatment in patients diagnosed with severe forms of HFM. Asymmetrical mandibular growth is also seen in patients who have suffered postnatal trauma or infection in the condylar region. This deformity differs from HFM in that it is limited to the jaw, without affecting the ear, soft tissues, or other organs. Therefore, mandibular asymmetry can be part of many conditions with different causes. According to the time of their onset, they might be related to (1) abnormality of early embryonic development (lack of neural crest cell migration) such as HFM or micrognathia (In these conditions, some alteration of growth patterns is observed as a consequence of the developmental abnormality. Usually orthopedic treatment has little probability of changing the pattern.) or (2) abnormality of late fetal or postnatal growth, where it is presumed that the abnormal process becomes causative after the embryonic period. This group includes abnormalities due to trauma, infection, or surgical iatrogenic deformities. Often these patients have involvement of only the bony structures of the mandible, but usually the soft tissues or the neuromuscular pattern is not affected. Therefore, these conditions are more likely to show good responses to functional stimulation.
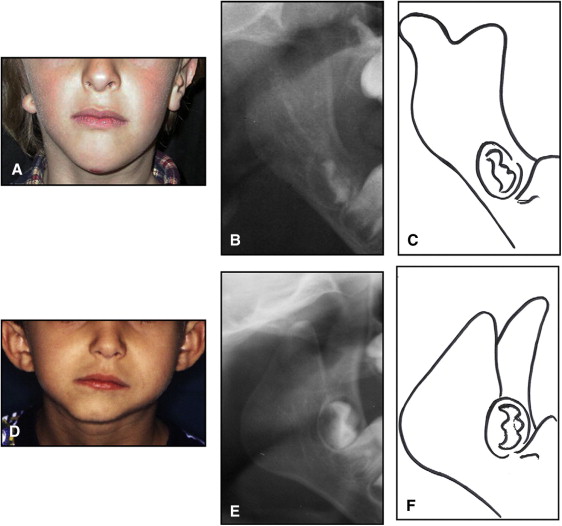
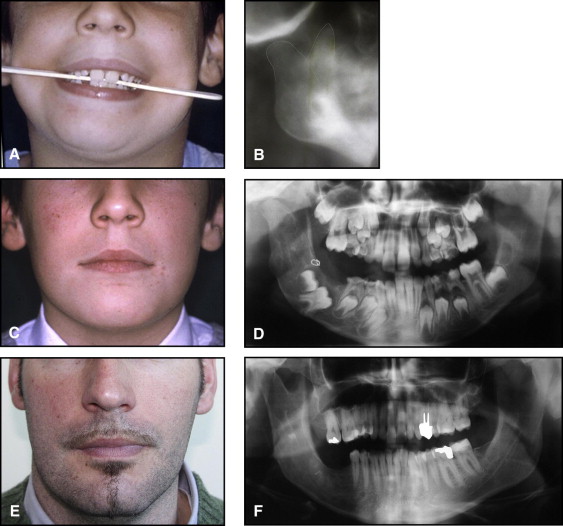
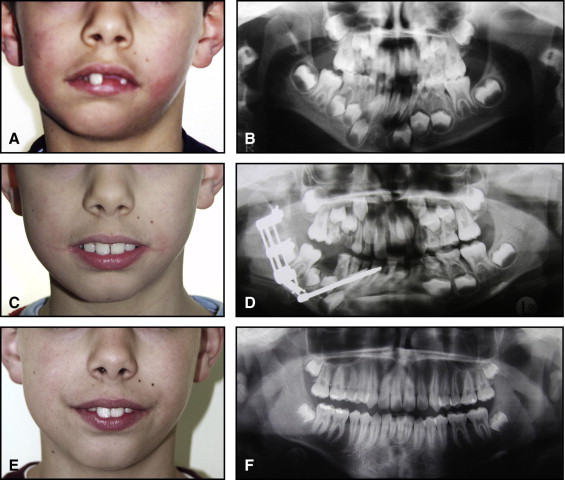
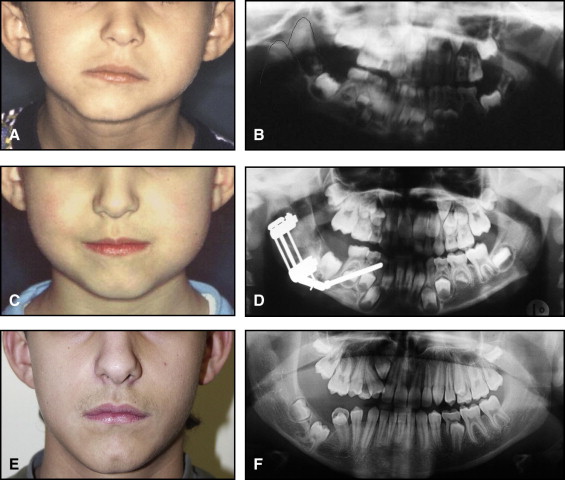
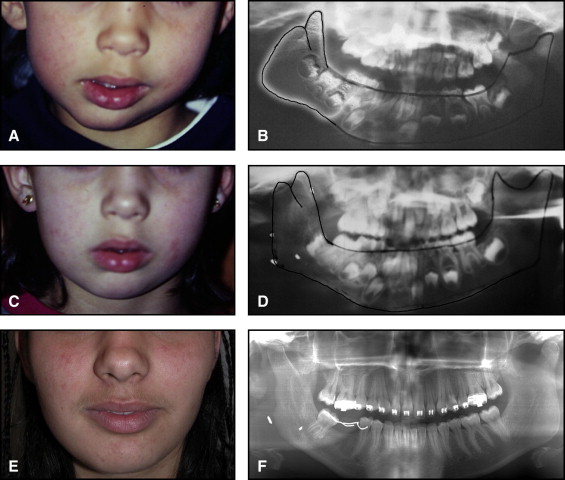
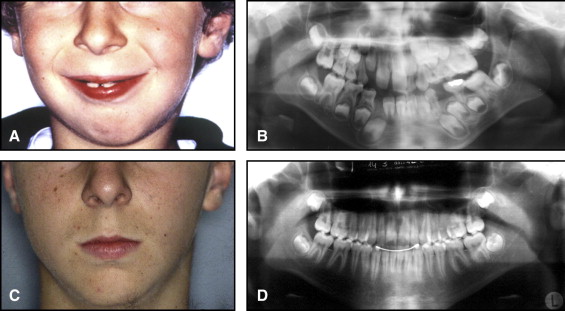
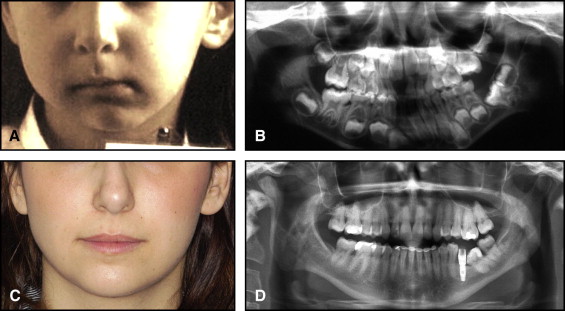
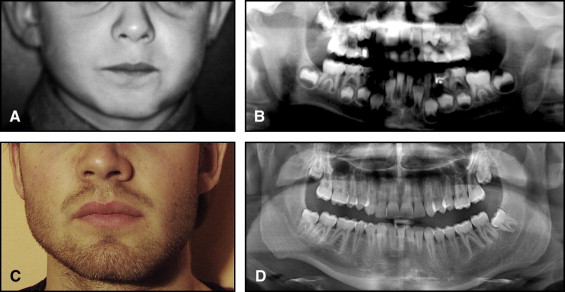
The aim of this article was to describe a peculiar type of mandibular asymmetry, frequently misdiagnosed as HFM. The patients shown share 2 main characteristics that distinguish them from more traditional HFM patients ( Table I , Fig 1 ). (1) There is no soft-tissue involvement, the external ear is present and well-formed, and the musculature seems to be well developed. Although the chin point deviates to the affected side, there is not the typical flatness of the gonial area seen in HFM patients ( Fig 1 , A-C ). On the contrary, there is more fullness on the affected side than on the unaffected side ( Fig 1 , D-F ). (2) The shape of the hypoplastic ramus is peculiar and extremely similar in all patients. The condyle is short and collapsed against the coronoid process ( Fig 1 , D-F ). Figures 2 through 5 show 4 patients who were treated surgically with excellent long-term results; Figures 6 through 10 show 5 patients who were treated orthodontically, with a similar remarkable ramal deformity correction. All had been erroneously diagnosed as having HFM by a surgeon or an orthodontist. Other patients with almost identical phenotypes misdiagnosed as having HFM can be found in the literature, and others already identified as without HFM can be also found.
| HFM | CCC | |
|---|---|---|
| History | Generally diagnosed at birth | Usually not diagnosed at birth Seldom history of trauma |
| Clinical examination | Soft-tissue defects (may be very mild) | No soft-tissue defects |
| Ear defects, preauricular tags | Normal ears, no preauricular tags | |
| Facial nerve asymmetries | No nerve deficit | |
| Masseter muscle hypoplasia | Well-developed masseter | |
| Deviation of the chin on the affected side, associated with flatness on the affected cheek | Deviation of the chin on the affected side, associated with fullness on the affected cheek | |
| Mild deviation to the affected side during opening | Significant deviation to the affected side during opening | |
| Panoramic x-ray (or computed tomograph) | Hypoplasia of the ramus and condyle and coronoid processes up to absence of the condyle and temporal fossa | Hypoplasia of the ramus and condyle and coronoid processes, which are typically collapsed one on the other; the temporal fossa is always present |
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


