Fig. 15.1
Intratemporal course of the facial nerve. The intratemporal course of the facial nerve consists of three segments: the labyrinthine, tympanic, and mastoid segments. The narrow bony confines of the labyrinthine segment combined with the absence of anastomosing arterial cascades make this segment most vulnerable to ischemic insult
From the stylomastoid foramen, the facial nerve passes between the digastric and stylohyoid muscles and enters the parotid. Branches supplying the posterior auricular muscles as well as the stylohyoid and posterior belly of the digastric exit the facial nerve before it enters the parotid. The facial nerve courses between the superficial and deep parotid lobes. At the pes anserinus, the facial nerve divides into the temporofacial and cervicofacial trunks. Further branching of the facial nerve results in the temporal, zygomatic, buccal, marginal mandibular, and cervical divisions ([69]; summarized in [65]). Many of these branches have variable branching patterns that may differ between the two sides of the face within the same individual. The facial nerve also demonstrates frequent interconnections among branches, resulting in redundant motor function [100], allowing transfer of a contralateral redundant branch for surgical reconstruction.
Pathology at any point along this pathway may result in facial nerve dysfunction. With knowledge of the facial nerve anatomy, a careful history and physical examination, occasionally combined with appropriate diagnostic studies, may identify the location of the nerve pathology and better guide management.
15.2 Etiopathology of Facial Paralysis
Facial paralysis (FP) may occur due to a multitude of processes, which are summarized in Table 15.1. The palsy may be complete or incomplete, congenital or acquired, bilateral or unilateral, and central or peripheral. Causes of facial nerve dysfunction differ between pediatric and adult patients, although Bell’s palsy is frequently causative in patients of all ages [16, 25, 80, 81, 102].
Table 15.1
Etiologies of facial paralysis
|
Congenital
|
Syndromic, non-syndromic, vascular malformation, Möbius syndrome, hemifacial microsomia, Goldenhar, Poland, Melkersson-Rosenthal (episodic)
|
|
Birth related
|
Traumatic or difficult delivery, instrumentation
|
|
Bell’s
|
Idiopathic, HSV
|
|
Traumatic
|
Temporal bone fracture, blunt force to cheek
|
|
Infectious
|
Acute otitis media, Lyme disease, VZV (Ramsay Hunt), HSV, EBV, mycoplasma, mastoiditis
|
|
Neoplastic
|
Central, parotid, or acoustic tumors
|
|
Iatrogenic
|
Brain, middle ear, and facial surgery
|
|
Ischemic
|
|
|
Neurogenic
|
Guillain-Barre
|
|
Hematologic
|
Leukemia, hemophilia
|
|
Hypertension
|
Bell’s palsy refers to idiopathic facial paralysis, which accounts for 30–70 % of reported cases of acquired facial palsy [7]. Herpes simplex virus (HSV) is now believed to account for this phenomenon. The virus is latent within the geniculate ganglion cells. At autopsy, polymerase chain reaction (PCR) has confirmed the presence of HSV genomic material in human geniculate ganglia [87]. In addition, facial paralysis has been induced by HSV infection in animal models [99], with a small percentage of animals exhibiting transient paralysis upon HSV reactivation [86]. Viral replication after reactivation causes facial nerve demyelination, resulting in neural edema. This edema may cause facial nerve compression within the fallopian canal, resulting in a cycle of neural damage (Fig. 15.2). While this phenomenon can occur at any age, the mean age of occurrence is 40–44 years (reviewed in [35]). A review of the emergency room visits for children with facial palsy in Taiwan determined that 50 % of cases resulted from Bell’s palsy [102].
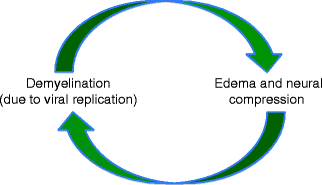
Fig. 15.2
Pathophysiology of Bell’s palsy. Herpes simplex viral replication results in nerve demyelination and edema which cause neural compression within the fallopian canal, creating a cycle of neural injury
In the pediatric population, Bell’s palsy, infectious etiologies, and trauma are the most common causes of facial palsy [16, 25, 80, 102]. In a review of 157 pediatric patients with facial palsy, Cha et al. [16] determined 66 % of cases resulted from Bell’s, while 14 % had infectious and 13 % traumatic etiologies. In their comparison to adult patients with facial palsy treated at the same institution, pediatric patients displayed higher rates of Bell’s palsy (66 % compared to 55 %) and traumatic (16 % compared to 5.9 %) etiologies. Shih et al. [80] described Bell’s in 78 % of their 56 cases of pediatric FP. A review of 35 cases of pediatric facial nerve palsy at Boston Children’s demonstrated 37 % of cases resulted from an infectious etiology, 20 % from trauma, and only 9 % from Bell’s. There was a bimodal distribution of ages of patients with traumatic and infectious causes of facial palsy at 2 years and 11.5 years [25]. In pediatric patients with an infectious etiology for FP, acute otitis media and varicella-zoster virus (VZV) [33, 67] are common instigators. Acute otitis media accounted for 10 % of the emergency room visits for FP in children, all under the age of 2 years, at Chang Gung Memorial Hospital [102]. Lyme disease is the most common cause of acute pediatric facial paralysis in endemic areas and should be considered in cases of bilateral paralysis and recent exposure to endemic areas in the summer months [81]. In general, the pediatric population with FP displays a greater incidence of trauma, neoplasm, and congenital anomalies compared to adult patients with FP [80, 102]. The facial nerve is positioned more laterally in infancy, making it more susceptible to trauma, including birth-related trauma [81]. When evaluating the pediatric patient with acquired facial paralysis, it is important to consider that facial palsy may be an early sign of systemic disease. Rarely, hematologic disorders, such as leukemia and hemophilia, and neoplastic processes may present with acute facial paralysis [81].
A knowledge of the etiology of facial paralysis provides valuable information regarding management, such as antibiotic and steroid administration and surgical intervention, but also may help delineate prognostic expectations. In the setting of Bell’s palsy, 10–15 % of patients regain no or poor function and 5–29 % have residual sequelae (such as spasm or synkinesis), which varies in severity [70, 71]. The overall recovery rate is related to the severity of paralysis. House-Brackmann grades of I or II often result in recovery rates of ∼90 % [16], and good recovery may be expected if neurodegeneration is <90 % [35]. Conversely, more severe paralysis with higher House-Brackmann scores carries a worse prognosis for recovery [49]. Age is inversely related to the recovery rate in patients with Bell’s palsy [22], and recovery is worse in patients greater than 50 years of age [49]. In pediatric patients with facial palsy, Evans et al. [25] noted faster recovery with infectious etiologies (∼1 month) compared to traumatic etiologies (∼8 months). Recovery rates range from 81 to 100 % for Bell’s palsy, 73–91 % for infectious etiologies, and 43–90 % in traumatic etiologies in the pediatric population [16, 25, 26, 83]. Prognosis is poorer in paralysis due to congenital and neoplastic processes [97, 102]. Adults demonstrate similar recovery rates of 89 % for infectious etiologies and 64 % from traumatic injury [16]. In general, prognosis is more favorable in cases with younger patient age, House-Brackmann score < III, normal nerve excitability testing, and a normal stapedial reflex [49, 84].
15.3 Evaluation
Careful evaluation of the patient with facial palsy is required to establish a proper diagnosis. Management of the patient with facial paralysis differs depending on the diagnosis. There is a fundamental difference in the natural history and pathology between acquired and congenital FP, for example. Acquired FP includes sequelae of reinnervation and may have a progressive or improving course. Congenital cases, however, may have absent or hypoplastic structures, a static course, and absence of reinnervation sequelae. Establishing the correct diagnosis for facial paralysis is essential to effective management.
A detailed history provides immense diagnostic information. The onset of symptoms should be discussed to determine the temporal pattern of onset. It is important to note that FP beginning from the time of birth does not automatically indicate a congenital etiology, as birth-related trauma, particularly use of instrumentation such as forceps, may result in facial nerve injury beginning at the time of birth. Birth history including birth weight, duration and difficulty of labor, and requirement for instrumentation can help to elucidate these different etiologies. In traumatic nerve injury, the mechanism of injury is important. The chronicity of onset is also important. An abrupt onset of FP may indicate an infectious, traumatic, or ischemic etiology, while an insidious, progressive onset of FP is more suggestive of a neoplastic process. The presence of any additional symptoms occurring simultaneously with the facial paralysis should also be noted. Simultaneous abducens nerve palsy suggests localization of the etiologic process to the region of the facial nucleus at the cerebellopontine angle, while hearing difficulties may indicate a process within the middle ear. For acquired cases of FP, the elapsed time from onset is important to determine potential for improvement and the possibility of reinnervation of native musculature.
The functional consequences of facial paralysis should also be elicited during initial evaluation. Protection of the cornea is paramount, particularly in acquired cases as patients with congenital FP tend to maintain corneal protection. The history should investigate frequent tearing, eye irritation, and the presence of lagophthalmos during sleep. Patients may describe difficulties with oral incompetence, speech (bilabial sounds), and altered taste sensation. Nasal breathing difficulties due to external valve collapse are often overlooked and should be discussed during the patient interview. The psychosocial impact of the patient’s FP should also be investigated.
A complete head and neck examination, including full cranial nerve evaluation, should be documented. Ocular examination should include assessment of extraocular movements, Bell’s reflex, and corneal sensation. Any abnormalities in corneal sensation necessitate ophthalmologic referral. Each facial nerve branch should be independently evaluated via brow elevation, forced eye closure, relaxed eye closure, maximal smile, lip elevation (show teeth), pucker, lower lip depression, and platysmal contraction. Facial symmetry is assessed both at rest and with animation. Particular note is made of midline shift of the mouth and asymmetries in oral commissure position at rest and with movement. Synkinetic and dyskinetic movements are also noted. Documentation of initial facial function at the time of presentation serves as an important baseline for outcomes assessment. The functional status of facial movements can be classified according to the House-Brackmann scale [48] (Tables 15.2 and 15.3). While this scale is not ideal for isolated paralysis of a facial nerve branch or congenital cases, it is user friendly and is the accepted scale of the Facial Nerve Disorders Committee of the American Academy of Otolaryngology—Head and Neck Surgery. In addition to grading, facial function should be documented photographically as well as with video with a standardized set of movements. Quantification of facial movements may also be assessed and documented in a number of ways ranging from measurements with a simple handheld ruler [4, 62] to complex videographic systems [32]. Bray et al. [14] have developed software that utilizes the standard reference of iris diameter to normalize smile measurements on still digital photographs [38, 41], providing an easy to use standard to determine oral commissure excursion that is universally applicable.
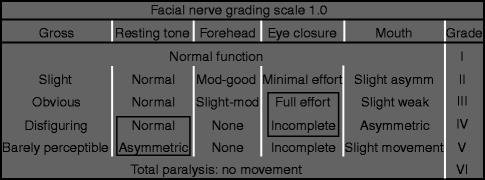
Table 15.2
The House-Brackmann facial nerve grading scale
|
Grade
|
Description
|
|---|---|
|
I
|
Normal
|
|
II
|
Mild dysfunction, slight weakness noticeable only on close inspection, good to moderate forehead function, complete eye closure with minimal effort, slight smile asymmetry with maximal effort, synkinesis barely noticeable
|
|
III
|
Moderate dysfunction, obvious but not disfiguring difference between sides, slight to moderate forehead movement, complete eye closure with effort, strong but asymmetric mouth movement, noticeable but not severe synkinesis
|
|
IV
|
Moderately severe dysfunction, obvious weakness and/or disfiguring asymmetry, normal rest symmetry and tone, inability to lift brow, incomplete eye closure, mouth asymmetry with maximal effort, severe synkinesis
|
|
V
|
Severe dysfunction, motion barely perceptible, rest asymmetry, no forehead motion, incomplete eye closure, slight movement corner mouth, synkinesis usually absent
|
|
VI
|
No movement, no synkinesis
|
Table 15.3
Modified House-Brackmann facial grading system
While not universally utilized, specific diagnostic studies may occasionally be useful adjuncts in the management of facial nerve injury. In the setting of acute FP, electroneuronography (ENOG) can predict which patients will have a poor outcome. ENOG records a compound action potential of facial muscle after a maximal electrically evoked transdermal stimulus near the stylomastoid foramen by measuring the movement of the facial muscles with a surface electrode at the nasolabial fold or perioral region. The recorded amplitude is proportional to the number of axon fibers that have a conduction block proximally at the level of the lesion that could still be stimulated (neurapraxia) [34]. Those axons that have undergone Wallerian degeneration (axonotmesis, neurotmesis) are unable to propagate electrically generated evoked potentials distal to the lesion. Results from the injured facial nerve are compared to the uninjured side, and deficits are expressed as a percentage of the action potential of the uninjured side. Patients with <90 % loss of function demonstrate satisfactory spontaneous recovery [35]. The timing of Wallerian degeneration may also be of prognostic value. Axons with a neurotmetic lesion undergo Wallerian degeneration early (3–5 days) and have no chance of recovery. An axonotmetic lesion will result in Wallerian degeneration later (14–21 days) and can still recover as regeneration occurs through the intact neural sheaths. Degeneration between 6 and 14 days will have variable degrees of axonotmesis versus neurotmesis and therefore recovery will be less predictable. ENOG is useful from days 3 to 21 after nerve injury/symptom onset. Beyond day 21, electromyography (EMG) is more useful [35, 81]. Fibrillations on EMG indicate neuronal injury, while motor unit potentials (MUPs) indicate recovery. EMG may also be beneficial to evaluate possible donor nerves in cases of multi-nerve dysfunction.
15.4 Treatment
The management goals of facial nerve injuries are to restore normal function, establish facial symmetry both at rest and with animation, minimize any donor deficit, and achieve normal facial contour through reliable and safe procedures. The pathway through which these goals are achieved varies depending upon the etiology and degree of facial paralysis, duration of denervation, and patient’s health, age, and goals. Multiple algorithms have been described to reflect this decision-making process [28, 35, 39, 65, 77, 89]. Regardless of the treatment pathway, corneal protection is of paramount importance and should be emphatically reviewed with patients.
Duration of denervation guides management. In cases of acquired facial nerve pathology, reinnervation of native facial musculature is ideal. After prolonged denervation, native musculature is no longer capable of reinnervation, and alternative reconstructive techniques must be employed. The duration of the acceptable denervation period has been somewhat debated but is generally agreed to be ∼12–18 months. Following nerve injury, Wallerian degeneration occurs distally. In this process, Schwann cells, fibroblasts, myocytes, and injured axons express neurotrophic factors as neural elements are degraded. In addition to the degeneration process, regeneration is also supported. After prolonged denervation (>6 months), however, distal Schwann cells provide less neuroregenerative support and may even undergo apoptosis [29, 76], providing a suboptimal environment for neuroregeneration. During denervation, changes occur not only in the nerve but also in the denervated muscle, a sequence of events termed denervation atrophy. Proteases cause myofibril reabsorption, myosin and actin filament catabolism, and increased collagen deposition [55]. Capillaries are lost, creating regions of avascular muscle [9]. Normally, acetylcholine receptors and neural cell adhesion molecules are upregulated to help guide neural ingrowth. Once the regenerating nerve reaches the target muscle, a neuromuscular junction is formed, and these growth factors are less ubiquitously expressed [55]. With increasing periods of denervation, the state of high affinity between the regenerating nerve and muscle diminishes. Eventually, the muscle becomes refractory to synaptic formation; it can never be reinnervated, regardless of neuroregeneration status. Motor reinnervation, then, is time-sensitive. Given the rate of neuroregeneration of 1 mm daily, lesions distant from end-target muscles may not be amenable to repair at the point of injury. Similarly, a cross-face nerve graft (CFNG) from the contralateral VIIth nerve may not be a viable solution for direct muscle reinnervation at 12 months post-injury. Nerve transfers provide a solution of providing motor input to the denervated muscle in a more timely manner as the transfer can be performed in closer proximity to the target than the nerve injury.
Facial nerve deficits are not always best managed surgically. In Bell’s palsy, improved recovery has been noted in patients receiving steroids and antivirals in close proximity to symptom onset [44, 85, 101]. Physiotherapy and biofeedback exercises are effective management tools for enhancement of facial movement and suppression of synkinesis. Neuromuscular reeducation is also imperative in the postoperative rehabilitation after nerve transfer procedures. Rehabilitation techniques are effective and continue to benefit patients even up to 3 years after facial nerve injury [57]. Chemodenervation also plays an important role in reducing synkinesis and improving facial symmetry [10, 30, 39, 53, 77]. The optimal treatment of facial nerve injuries requires a multimodality approach [39], and the importance of these techniques should not be underestimated, although they are not the focus of this chapter and are not thoroughly discussed.
15.4.1 Surgical Management of Acute Facial Nerve Lesions
Direct facial nerve repair should be completed when possible. Nerve repair within 72 h from the time of injury allows stimulation to aid identification of distal nerve ends [103]. After 72 h from injury, motor end-plate depolarization is not possible as neurotransmitter stores are depleted in the absence of antegrade axoplasmic transport. Repairs should always be performed without tension. If a tensionless repair is not possible, nerve autografts should be utilized. Common donors are the sural, great auricular, cervical plexus, medial antebrachial cutaneous, and lateral antebrachial cutaneous nerves. The neurorrhaphy should be performed with healthy nerve ends. An elegant nerve repair or reconstruction performed within the zone of injury has no functional value. Facial nerve repair should be completed with meticulous technique, by surgeons with experience in neurorrhaphy, in an appropriate setting with proper lighting, magnification, and instruments.
Depending upon the mechanism of injury, the facial nerve may be in continuity but incur injury due to external compression. Neural edema within the bony confines of the intratemporal segment can impair function. Facial nerve decompression was first described as a treatment for Bell’s palsy by Balance and Duel in 1932 [6] and is controversial. Proponents claim the usefulness of decompression when performed within 12–14 days of traumatic injury or the onset of Bell’s palsy in patients who will have a poor recovery or who have an unfavorable electrodiagnostic profile [35, 46]. Poorer recovery has been reported when decompression was performed more than 2 weeks after nerve injury [46]. Multiple potential sites of compression exist, although differences in functional recovery by region of decompression have not been shown [46]. Opponents of facial nerve decompression also cite the difficulty in predicting which patients will have a poor outcome, although this task is aided with ENOG, and note few differences in recovery for patients who have undergone decompression compared to those with spontaneous recovery [1]. The authors have not utilized this procedure in the management of facial nerve injuries.
In the setting of acute facial nerve injuries when a proximal facial nerve stump is not available (damaged in trauma, resected with tumor, etc), an alternative neuronal source should be used to reinnervate the native mimetic musculature. The contralateral facial nerve or other nearby cranial nerves can provide neuronal input.
If available, the contralateral facial nerve in combination with a cross-face nerve graft (CFNG) (typically the sural nerve) may be used to innervate the affected facial muscles. First described by several authors in the early 1970s [2, 78, 82], this technique has the benefit of borrowing nerves of like function to reconstruct facial movement and therefore allows spontaneous and emotional facial expression without dyskinesis. In addition, the need for motor reeducation is eliminated. Due to redundancy of facial nerve branches and complex facial muscle innervation patterns, a branch of the contralateral facial nerve can be harvested after careful facial nerve mapping with minimal to no donor deficit. The downside of this technique is that the innervation source is not as robust as some other cranial nerves, such as branches of the trigeminal. The reconstruction requires neuroregeneration across a lengthy CFNG and two coaptation sites. The increased time for neuroregeneration allows greater muscle atrophy and a poorer neuronal trophic environment [52]. Through a preauricular incision with caudal extension just posterior to the mandible on the unaffected side of the face, the healthy facial nerve branches are meticulously mapped. For smile reconstruction, a buccal or zygomatic branch is identified, typically at a point half the distance between the tragus and the oral commissure (Zuker’s point). Nerve branches are identified and then stimulated to determine function. The branch that provides oral commissure, upper lip, and alar elevation, with redundant function, is selected as the donor. Use of a donor branch that produces the intended motion may decrease synkinesis and involuntary motion as the donor has similar cortical origins as the injured side. Care is taken to ensure redundancy and preservation of periocular muscle function. The injured facial nerve segment is exposed through an identical preauricular incision on the injured side of the face. A subcutaneous tunnel is created in the upper lip, and a nerve graft is passed between the two sides of the face. Tensionless neural coaptations are then performed between the donor facial nerve and the nerve graft and between the downstream nerve graft and the distal segment of the injured facial nerve. Return of facial nerve function is often noted 6–9 months after reconstruction. Neuroregeneration may be followed by an advancing Tinel’s sign across the CFNG. This technique may also be performed in two stages: donor branch selection and coaptation with CFNG, followed by coaptation of the CFNG to the distal target nerves in a second procedure approximately 6–12 months later.
If the contralateral facial nerve is unavailable and the native facial musculature remains appropriate for reinnervation, an alternative neural source may be utilized for direct muscle reinnervation. The ipsilateral masseter branch of the trigeminal nerve provides a robust axonal source, often without the need for an intervening nerve graft and therefore with only a single coaptation. Use of this neuronal source was initially described by Zuker et al. [108] for free segmental muscle transplantation to the face for patients with Möbius syndrome, but this nerve transfer can also be used for direct mimetic muscle neurotization [21]. The motor nerve to the masseter muscle can be identified along the deep surface of the masseter muscle coursing obliquely from the posterior-superior to the anterior-inferior muscle borders [51, 108]. It is located approximately 3 cm anterior to the tragus and 1 cm inferior to the zygomatic arch [11]. Because of the close proximity between the facial and trigeminal nuclei in the pons, motor reeducation after this procedure is often rapid, even in the absence of formal therapy, for both adult and pediatric patients. The ability to smile without active biting has been reported in 66–82 % of patients [51, 61, 75, 105]. The motor branch to the masseter provides a robust innervation source with greater axonal numbers compared to the buccal branch of the facial nerve, with clinical facial movement often noted 3 months postoperatively [21]. No donor deficit has been reported after use of the motor branch to the masseter [21, 61, 108].
If both the contralateral VII and the motor branch to the masseter are unavailable for mimetic muscle reinnervation, alternative nerve transfers such as the spinal accessory (first completed by Drobnick in 1879, as cited by Griebie [37]), hypoglossal [20], partial spinal accessory [13], partial hypoglossal [3, 64], phrenic [73, 106], and C7 [90] nerves have been successfully used. These alternative nerve sources, however, have increased donor morbidity and often result in dyskinetic facial motions (reviewed in [72]). Today, none of these procedures are considered first-line donor nerve selections by our group.
The babysitter technique, described by Terzis [88, 95], utilizes the concept of reinnervation while waiting for definitive neuroregeneration with use of a partial hypoglossal nerve transfer. The theory is that immediate reinnervation will protect motor end plates within the target muscle and prevent denervation atrophy. The question of whether two separate reinnervation procedures are more harmful than a single denervation period followed by a single reinnervation transition has been raised in the literature [107].
15.4.2 Surgical Management of Established Facial Nerve Lesions
With prolonged muscle denervation (approximately 12–24 months), changes within both the target nerve and muscle preclude further reinnervation of native musculature. As a result, surgical intervention may establish static or dynamic reconstructions depending upon associated patient factors, such as physical health, psychological state, psychosocial support, and preference.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


