Hematopoietic cell transplantation is used to treat malignancies, hematologic and immune deficiency states, marrow failure syndromes, and autoimmune diseases. Graft-versus-host disease (GVHD) is a clinical syndrome seen following allogeneic transplantation where donorderived immunocompetent T cells and inflammatory responses attack host tissues. GVHD can cause significant morbidity and even result in mortality. The oral cavity is a frequently involved site with clinical changes resembling autoimmune collagen vascular diseases. Recognition, diagnosis, and monitoring of oral GVHD can help with diagnosis and grading of GVHD and judging responses to therapy. Topical and local management of symptomatic oral GVHD can reduce oral symptoms that can interfere with oral function and quality of life, and can reduce the need for more intensive immunosuppressive systemic therapies.
Hematopoietic cell transplantation (HCT) began in the late 1950s and has seen a steady increase in the overall success and applicability to treat a wide range of malignancies, hematologic and immune deficiency states, and autoimmune diseases . Animal studies in the late 1940s and early 1950s laid the ground work for HCT, and human HCT was first attempted in the late 1950s. However, early HCT attempts were complicated by significant and often lethal complications including infection, graft failure, relapse, hemorrhage, and, in the case of allogeneic HCT (alloHCT), graft-versus-host disease (GVHD) . GVHD is a clinical syndrome where donor-derived immunocompetent T cells react against patient tissues directly or through exaggerated inflammatory responses following alloHCT . The primary target organs of GVHD classically have been those of skin, liver, and the gastrointestinal (GI) tract. However, the oral cavity is also frequently involved, possibly only second to cutaneous involvement . Despite significant advances, GVHD remains a major cause of morbidity and mortality with chronic GVHD being the leading cause of nonmalignant fatality post alloHCT .
Given the impact of GVHD on the allogeneic patient’s post HCT course, timely and accurate diagnosis of oral GVHD, ongoing assessment of responses to therapy, and the appropriate management of oral GVHD can contribute to not only improved patient comfort, oral health, and function, but possibly long-term survival . Since the oral cavity is frequently involved and is easily assessed for GVHD, recognition of oral GVHD changes can contribute to improved post-HCT medical management. Finally, appropriate therapy for symptomatic oral GVHD can significantly improve the patient’s quality of life and overall oral function.
Pathobiology, epidemiology, and clinical manifestations of graft-versus-host disease
In 1966 Billingham formulated the three fundamental elements required for the occurrence of GVHD. First, the transplanted graft must contain immunologically competent cells; second, the recipient must be incapable of rejecting the transplanted cells; and third, the recipient must express tissue antigens that are not present in the donor . In the most simplistic terms, based on these fundamental principles, GVHD is a clinicopathologic syndrome that occurs following alloHCT when transplanted immunologically competent donor (graft) T cells recognize and react against histocompatability antigens on the patient’s cells (host) and induce immune responses resulting in host tissue damage . These processes are triggered by the recognition of host human leukocyte antigens (HLAs) by donor lymphocytes as being “foreign” antigens; the ensuing complex set of both autoimmune and alloimmune responses and severe inflammatory manifestations are recognized clinically as GVHD .
The most important risk factor for GVHD is the degree of HLA match of donor to patient. Mismatching in HLA-A, -B, -C, or -DRB1 will increase the risk of GVHD . Sibling HLA-matched (ie, related) donor grafts have less risk for GVHD than HLA-matched unrelated donor grafts, possibly because of mismatched minor histocompatibility antigens (mHA) that have been shown to influence GVHD incidence. Further immune-based differences that influence the risk for GVHD are being studied . Additional risk factors for GVHD include increasing patient age, donor parity and sex mismatch, choice of graft source (ie, peripheral blood stem cells, bone marrow, or umbilical cord), and pre-infusion graft modulations/manipulations, most notably T-cell depletion . Finally, the toxicity of high-intensity conditioning regimens, especially those that use total body irradiation, can increase GVHD risk because they cause more host tissue damage that results in enhanced recognition of host antigens by donor antigen-presenting cells (APCs) leading to increased activation of donor T cells.
GVHD was initially classified as either acute GVHD (aGVHD) or chronic GVHD (cGVHD) based on an arbitrary time point with aGVHD occurring within 100 days of transplantation and cGVHD occurring over 100 days after transplantation. Recently, there has been a shift toward separation of acute and chronic forms of GVHD based on clinical and pathological characteristics . While both forms represent the consequence of damage to host tissues by activated donor-derived T lymphocytes in response to the major histocompatibility complex (MHC) disparities between the donor and the host, elucidation of the immunopathobiology of GVHD has made it apparent that the specific pathophysiological mechanisms are distinctly different . Acute GVHD can occur as early as 1 week post-HCT or following donor lymphocyte infusion (DLI, see next paragraph). In contrast cGVHD can have an onset of 70 days or later post-HCT or DLI and continue for many years. There are four patterns of onset for cGVHD: (1) a de novo onset (without prior aGVHD), (2) quiescent onset (onset is after a period of no apparent aGVHD activity between the resolution of aGVHD and the onset of cGVHD), (3) progressive onset (cGVHD evolves directly from aGVHD), and (4) explosive onset (manifesting with an abrupt onset of severe multisystem involvement with manifestations of both acute and chronic GVHD) .
While both forms of GVHD contribute to significant post-HCT morbidity and mortality, for patients transplanted for malignancies, GVHD has also been associated with lower relapse rates because of a graft-versus-leukemia (GVL or graft-versus-tumor, GVT) effect . Chronic GVHD in particular has been shown to be associated with lower relapse rates, especially for patients who are transplanted in relapse . The GVL effect is thought to be mediated, at least in part, by donor T cells; however, the precise mechanisms that result in GVHD and GVL remain to be elucidated. Recognition of this beneficial effect has led to the use of a technique for patients who relapse post-HCT in which lymphocytes are collected from the donor and infused into the patient (referred to as a DLI). The goal of this treatment is to induce a T-cell–mediated GVL effect and has proven to be successful in many instances . Unfortunately, DLIs will also induce GVHD with its associated morbidity and mortality.
Acute graft-versus-host disease
Acute GVHD has an incidence rate of approximately 20% to 48% for matched related-donor HCTs, up to 70% for matched unrelated-donor HCTs, and MHC-mismatched (HLA haploidentical) patients have rates as high as 80% to 90% . If related donors are mismatched for 1-, 2-, or 3- HLA antigens, the risk of GVHD ranges from 75% to 80% . Acute GVHD generally occurs within 14 to 35 days of stem cell infusion but can be seen as early as within 1 week of transplant . As noted above, the incidence and severity of aGVHD is related to several immunologically based factors, such as donor-recipient HLA disparity, donor-recipient gender differences, source of stem cells, number of T cells in donor stem cell infusion, and effectiveness of GVHD prophylaxis regime . In patients receiving conventional GVHD prophylaxis, such as a combination of cyclosporine and methotrexate, the median onset of GVHD is 21 to 25 days after HCT, however onset may be delayed in T-cell depleted grafts . Reduced intensity conditioning regimens (previously referred to as nonmyeloablative conditioning regimes) may further decrease the incidence of aGVHD . A hyperacute form of GVHD may occur in patients with severe HLA mismatches and in patients who receive inadequate GVHD prophylaxis . Hyperacute GVHD is a severe form of aGVHD that can occur in the first 1 to 2 weeks after HCT and can be rapidly fatal .
The pathobiology of aGVHD has been described as a three-step process in which the innate and adaptive immune systems interact: (1) tissue damage to the recipient occurs from the radiation/chemotherapy conditioning regimen; (2) donor T cells recognize host antigens from the damaged tissues as foreign and become activated and stimulated, and then clonally expand; and (3) an effector stage ensues that is characterized by damage to host tissues induced either directly by immune cells or through a series of complex immune and inflammatory responses. In step one, conditioning regimens directly damage host tissues releasing alloantigens and inflammatory cytokines that promote activation of host APCs . In step two, host APCs present the host’s alloantigens to the resting donor T cells, thus activating them. Donor T-cell activation is characterized by cellular proliferation and the production of inflammatory cytokines, including interleukin (IL)-2, tumor necrosis factor alpha (TNF-α), and interferon gamma (INF-γ). Step three is characterized by multiple cytotoxic effectors including inflammatory cytokines, cytotoxic T cells, natural killer cells, other cells (mononuclear phagocytes and neutrophils), and nitric oxide production, producing direct target tissue damage or through intense inflammation associated with intense cytokine production, the so-called “GVHD cytokine storm” . The initial inflammation results in the further recruitment of effectors cells into target organs, amplifying local tissue injury with further secretion of inflammatory cytokines, which, together with cytotoxic T lymphocytes, lead to target tissue destruction . Additionally, specific proliferating marrow cells (eg, NK1.1 + T-cells and NK1.1 – T cells) are capable of suppressing or promoting T-cell responses and thus modulate the incidence and severity of aGVHD . Interestingly enough, donor-host histocompatibility differences may not always be needed to produce GVHD. A GVHD-like syndrome, seen rarely following autologous HCT, appears to arise through the inappropriate recognition of self-antigens . This is usually a mild self-limiting condition that readily responds to treatment with steroids.
The classically reported target organs for clinical manifestation of aGVHD are skin, liver, and the GI tract, but oral manifestations have clearly been documented . In the skin, a pruritic skin rash is noted with generalized erythoderma and in severe cases bullae formation and desquamation. Damage to the GI tract from conditioning regimen toxicity increases the translocation of inflammatory stimuli such as endotoxins, which promotes further inflammation and additional damage. Signs and symptoms of GI aGVHD include nausea, vomiting, diarrhea, and pain. Hepatic aGVHD is characterized as a cholestatic jaundice with a marked rise in bilirubin levels. Acute GVHD may also affect hematopoiesis resulting in a reduction in peripheral blood counts, particularly platelets .
Chronic graft-versus-host disease
cGVHD is the most common late complication after alloHCT and is the leading cause of late HCT non–relapse-related mortality . cGVHD prevalence varies from 25% to 80% in long-term survivors after alloHCT with 5-year survival rates as low as 40% for patients with severe multisystem cGVHD . The incidence and severity of cGVHD are correlated to immunologically related factors including HLA disparity, donor/host age and sex, donor type, source of progenitor cells, graft manipulations (especially T cell depletion), previous aGVHD, and use of post-HCT DLIs . Often, cGVHD will present during tapering of or soon after stopping aGVHD prophylaxis or treatment .
The pathophysiology of cGVHD is not well understood. There are two theories regarding the mechanism of cGVHD: the first is simply end-stage alloreactivity and the second is that cGVHD is caused by poor/dysfunctional immunologic recovery with the evolution of autoreactive T lymphocytes because of lack of thymic control . cGVHD is a multisystem alloimmune and autoimmune disorder characterized by immune dysregulation, immunodeficiency, and impaired organ function, all of which negatively impact survival . The pathobiology of cGVHD starts with the expansion of donor T cells in response to alloantigens or autoantigens that is unchecked by normal thymic or peripheral mechanisms of deletion. T cells promote target organ damage either directly through inflammatory cytokines, cytolytic attack and fibrosis, and/or by promoting B-cell activation and production of autoantibodies . While there can be widespread organ damage, the leading cause of death for patients with cGVHD is infection .
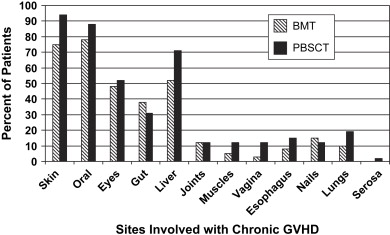
The most common sites of cGVHD involvement are the skin, oral cavity, eyes, GI tract, liver, and lungs ( Fig. 1 ); however, the spectrum of clinical involvement is remarkably variable . Clinical features are similar to those of collagen vascular diseases, lichen planus (LP), Sjögren syndrome, polyserositis, esophagitis and stricture, vaginal ulcerations and stricture, intrahepatic obstructive liver disease, obstructive pulmonary disease, progressive systemic sclerosis, fasciitis, and myositis .
Oral graft-versus-host disease
Oral acute graft-versus-host disease
Epidemiological studies of oral aGVHD are lacking, but anecdotally we estimate that probably between 35% and 60% of patients with aGVHD will have oral manifestations. Oral aGVHD can occur as early 1 to 2 weeks post-HCT but more often between 18 and 100 days post-HCT; however, early onset of oral aGVHD is usually obscured by (or mistaken for) conditioning regimen–related mucositis . Oral mucosal involvement has not been part of the classic description of aGVHD. However, if infection is excluded with appropriate cultures, ulcerative lesions that fail to heal with hematologic recovery 21 to 28 days post-HCT may well represent oral aGVHD, although this does not become clinically distinct from oral mucositis until almost 3–4 weeks post-HCT .
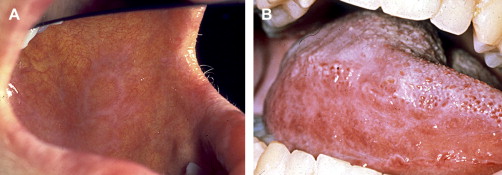
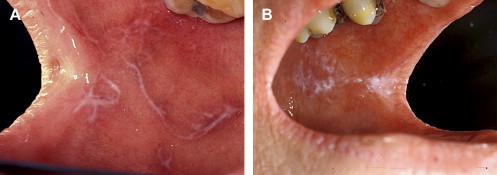
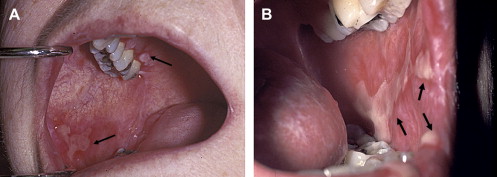
Clinically, acute and chronic oral GVHD are characterized by mucosal hyperkeratotic responses, erythema and inflammation, atrophy, pseudomembranous ulcerations, fibrosis, and salivary gland dysfunction and taste disorders in patterns reminiscent of autoimmune disorders such as LP, lupus, systemic sclerosis, and Sjögren syndrome ( Figs. 2–6 ). The clinical manifestations and histopathology of oral aGVHD and cGVHD also are very similar, although aGVHD changes tend to be less pronounced and distinct, and tends to be predominated more by erythema and atrophy, especially before day 50 post-HCT; however, LP-like hyperkeratotic changes can be noted but are less prominent than what is generally noted for cGVHD (see Fig. 2 ). Pseudomembranous ulcerations are noted across the spectrum of aGVHD and cGVHD and are an indicator of severity (see Fig. 4 ). Whether these variations are due specifically to different pathologic mechanisms or a result of the effect of chronicity and severity of attack is not known.
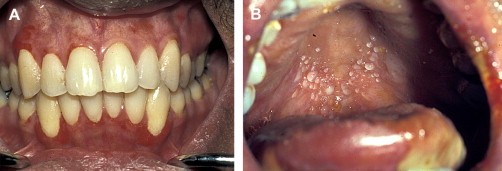
Oral chronic graft-versus-host disease
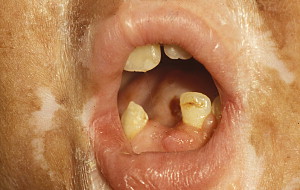
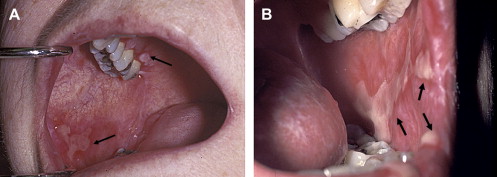
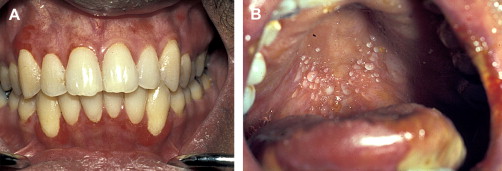
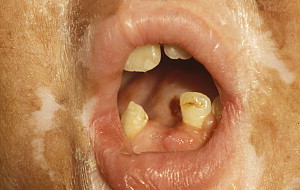
In contrast to oral aGVHD, the presence of oral cGVHD is a frequent, prominent, and useful component of cGVHD diagnosis and staging. The frequency of oral cGVHD has been well documented with 72% to 83% of patients with cGVHD showing oral involvement, making this one of the most common manifestations of cGVHD (see Fig. 1 ) . Clinical changes include mucosal inflammation and atrophy, lichenoid-hyperkeratotic changes (striae, plaques, papules, and patches), pseudomembranous ulcerations, mucoceles, and perioral fibrosis (see Figs. 3–6 ). Clinically, cGVHD oral mucosal inflammation tends to produce a vasculitis-like or even a telangiectatic appearance, especially with longer standing involvement. Chronic GVHD involvement of the maxillary anterior attached gingival is characterized by atrophy with loss of stippling and moderate or worse inflammation of the entire attached gingiva; LP-like changes also can be noted. Sclerotic changes in the perioral tissues may result in decreased oral opening ( Fig. 6 ). The symptoms most often reported are pain (usually associated with ulcerative changes), sensitivity to normally tolerated items (hard crunchy foods, spices, acidic and carbonated beverages, and mint and cinnamon flavoring), xerostomia, and occasionally taste dysfunction . Based on the characteristic oral changes and symptoms, the National Institutes of Health (NIH) Consensus Development Project for Clinical Trials in Chronic GVHD has developed a scoring system for following established oral cGVHD ( http://www.asbmt.org/GvHDForms ) .
Salivary gland chronic graft-versus-host disease
Saliva plays a major role in maintaining oral health and oral function . When salivary function is compromised there is a significant increase in oral complications including increased risk for dental caries; mucosal infections; mucosal pain and friability; and difficulties with speaking, chewing, and swallowing . The effects of HCT, and especially cGVHD, on salivary gland secretions have been extensively studied in both animal models and clinically. Early post-alloHCT salivary gland dysfunction is primarily attributed to conditioning regimen toxicity, especially when total body irradiation is used, and can persist for many months; however, late changes, especially in the context of systemic involvement (eg, skin) can be attributed to cGVHD . Sialochemical and sialometric changes associated with cGVHD have been extensively reported . Severe involvement results in the total destruction of secretory units and thus permanent and profound oral dryness .
Taste dysfunction
While conditioning regimen–related ageusia or dysgeusia is a multifactorial phenomenon that typically resolves 1 to 2 months following HCT , late and selective taste disorders can persist for over 100 days post-HCT . Patients may report a rapid decrease in their sense of taste that is temporally associated with the onset (or flare) of cGVHD, which suggests that the epithelial-derived taste receptor cell may be an immune-based target . It is important to note, however, that calcineurin inhibitors (cyclosporine, tacrolimus) used to prevent and treat GVHD also can induce neurological changes that result in dysgeusia or ageusia, and this can confound the interpretation of this symptom relative to GVHD.
Histopathological findings
The diagnosis of oral cGVHD can frequently be made based on clinical presentation. When oral changes are less distinct, biopsy of the oral mucosa and minor salivary glands can provide important supportive diagnostic information .Oral mucosal and salivary gland biopsy changes are initially dominated by conditioning regimen–related toxicity, but by 50 to 60 days post-alloHCT, changes attributable to GVHD are more apparent . In general, histologically there is little distinction between acute and chronic GVHD and, given the possibility for nonspecific inflammation, early post-HCT biopsy findings need to be correlated with clinical changes to confirm the diagnosis . Features include lichenoid interface inflammation, exocytosis, and keratinocyte apoptosis ( Fig. 7 ). Immunohistopathological studies demonstrate the presence of CD8-positive T cells, CD68-positive macrophages, and Langerhans cells, although B cells may also play a role .
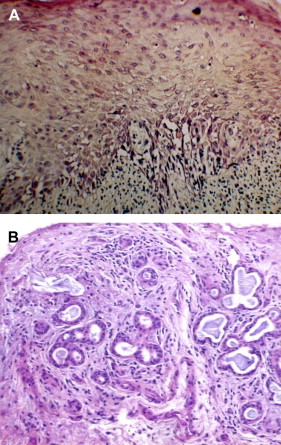
Salivary gland changes are characterized by the presence of lymphocytic infiltration (CD8 + over CD4 + ) of the glandular parenchyma and periductal tissue with or without plasma cells and exocytosis of lymphocytes (without neutrophils) into intralobular ducts and acini (see Fig. 7 ) . The damage to salivary acini ultimately leads to acinar destruction with fibrosis. Superficial mucoceles are likely a result of GVHD-induced inflammation and damage to the minor salivary gland ducts with initial duct obstruction and later destruction while the acini are still functional.
Oral graft-versus-host disease
Oral acute graft-versus-host disease
Epidemiological studies of oral aGVHD are lacking, but anecdotally we estimate that probably between 35% and 60% of patients with aGVHD will have oral manifestations. Oral aGVHD can occur as early 1 to 2 weeks post-HCT but more often between 18 and 100 days post-HCT; however, early onset of oral aGVHD is usually obscured by (or mistaken for) conditioning regimen–related mucositis . Oral mucosal involvement has not been part of the classic description of aGVHD. However, if infection is excluded with appropriate cultures, ulcerative lesions that fail to heal with hematologic recovery 21 to 28 days post-HCT may well represent oral aGVHD, although this does not become clinically distinct from oral mucositis until almost 3–4 weeks post-HCT .
Clinically, acute and chronic oral GVHD are characterized by mucosal hyperkeratotic responses, erythema and inflammation, atrophy, pseudomembranous ulcerations, fibrosis, and salivary gland dysfunction and taste disorders in patterns reminiscent of autoimmune disorders such as LP, lupus, systemic sclerosis, and Sjögren syndrome ( Figs. 2–6 ). The clinical manifestations and histopathology of oral aGVHD and cGVHD also are very similar, although aGVHD changes tend to be less pronounced and distinct, and tends to be predominated more by erythema and atrophy, especially before day 50 post-HCT; however, LP-like hyperkeratotic changes can be noted but are less prominent than what is generally noted for cGVHD (see Fig. 2 ). Pseudomembranous ulcerations are noted across the spectrum of aGVHD and cGVHD and are an indicator of severity (see Fig. 4 ). Whether these variations are due specifically to different pathologic mechanisms or a result of the effect of chronicity and severity of attack is not known.
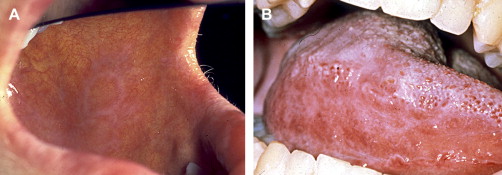
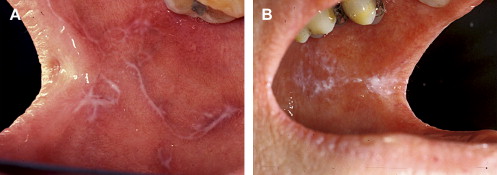
Oral chronic graft-versus-host disease
In contrast to oral aGVHD, the presence of oral cGVHD is a frequent, prominent, and useful component of cGVHD diagnosis and staging. The frequency of oral cGVHD has been well documented with 72% to 83% of patients with cGVHD showing oral involvement, making this one of the most common manifestations of cGVHD (see Fig. 1 ) . Clinical changes include mucosal inflammation and atrophy, lichenoid-hyperkeratotic changes (striae, plaques, papules, and patches), pseudomembranous ulcerations, mucoceles, and perioral fibrosis (see Figs. 3–6 ). Clinically, cGVHD oral mucosal inflammation tends to produce a vasculitis-like or even a telangiectatic appearance, especially with longer standing involvement. Chronic GVHD involvement of the maxillary anterior attached gingival is characterized by atrophy with loss of stippling and moderate or worse inflammation of the entire attached gingiva; LP-like changes also can be noted. Sclerotic changes in the perioral tissues may result in decreased oral opening ( Fig. 6 ). The symptoms most often reported are pain (usually associated with ulcerative changes), sensitivity to normally tolerated items (hard crunchy foods, spices, acidic and carbonated beverages, and mint and cinnamon flavoring), xerostomia, and occasionally taste dysfunction . Based on the characteristic oral changes and symptoms, the National Institutes of Health (NIH) Consensus Development Project for Clinical Trials in Chronic GVHD has developed a scoring system for following established oral cGVHD ( http://www.asbmt.org/GvHDForms ) .
Salivary gland chronic graft-versus-host disease
Saliva plays a major role in maintaining oral health and oral function . When salivary function is compromised there is a significant increase in oral complications including increased risk for dental caries; mucosal infections; mucosal pain and friability; and difficulties with speaking, chewing, and swallowing . The effects of HCT, and especially cGVHD, on salivary gland secretions have been extensively studied in both animal models and clinically. Early post-alloHCT salivary gland dysfunction is primarily attributed to conditioning regimen toxicity, especially when total body irradiation is used, and can persist for many months; however, late changes, especially in the context of systemic involvement (eg, skin) can be attributed to cGVHD . Sialochemical and sialometric changes associated with cGVHD have been extensively reported . Severe involvement results in the total destruction of secretory units and thus permanent and profound oral dryness .
Taste dysfunction
While conditioning regimen–related ageusia or dysgeusia is a multifactorial phenomenon that typically resolves 1 to 2 months following HCT , late and selective taste disorders can persist for over 100 days post-HCT . Patients may report a rapid decrease in their sense of taste that is temporally associated with the onset (or flare) of cGVHD, which suggests that the epithelial-derived taste receptor cell may be an immune-based target . It is important to note, however, that calcineurin inhibitors (cyclosporine, tacrolimus) used to prevent and treat GVHD also can induce neurological changes that result in dysgeusia or ageusia, and this can confound the interpretation of this symptom relative to GVHD.
Histopathological findings
The diagnosis of oral cGVHD can frequently be made based on clinical presentation. When oral changes are less distinct, biopsy of the oral mucosa and minor salivary glands can provide important supportive diagnostic information .Oral mucosal and salivary gland biopsy changes are initially dominated by conditioning regimen–related toxicity, but by 50 to 60 days post-alloHCT, changes attributable to GVHD are more apparent . In general, histologically there is little distinction between acute and chronic GVHD and, given the possibility for nonspecific inflammation, early post-HCT biopsy findings need to be correlated with clinical changes to confirm the diagnosis . Features include lichenoid interface inflammation, exocytosis, and keratinocyte apoptosis ( Fig. 7 ). Immunohistopathological studies demonstrate the presence of CD8-positive T cells, CD68-positive macrophages, and Langerhans cells, although B cells may also play a role .
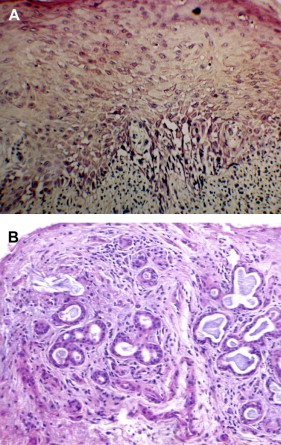
Salivary gland changes are characterized by the presence of lymphocytic infiltration (CD8 + over CD4 + ) of the glandular parenchyma and periductal tissue with or without plasma cells and exocytosis of lymphocytes (without neutrophils) into intralobular ducts and acini (see Fig. 7 ) . The damage to salivary acini ultimately leads to acinar destruction with fibrosis. Superficial mucoceles are likely a result of GVHD-induced inflammation and damage to the minor salivary gland ducts with initial duct obstruction and later destruction while the acini are still functional.
Impact of graft-versus-host disease
The impact of GVHD on patients depends on the severity of the disease and the effectiveness of treatment protocols . When severe, both acute and chronic extensive GVHD have to be considered a potentially life-threatening condition. Because infection is the leading cause of nonrelapse mortality in patients with cGVHD, infectious disease prophylaxis is critical, as is ensuring rapid diagnosis and treatment of infectious complications that develop. Supportive care must be directed toward managing the deleterious effects of cGVHD involvement (eg, hepatic, pulmonary, dermal, ocular, hematopoietic, nervous, and musculoskeletal) and the side effects of therapies, while maintaining an adequate quality of life . Additionally, it is important to educate patients; refer for nutritional counseling; and to use active interventions to manage the financial, physical, emotional, and psychological complications of cGVHD .
The impact of oral involvement depends on its severity and duration and ranges from inconsequential to devastating . Oral and dental supportive care measures aimed at managing mucosal and salivary gland disease play a major role in post-alloHCT patient care (see the following section) . When symptomatic, oral GVHD can affect the patient’s quality of life because of associated pain, sensitivity, and dryness, and can significantly interfere with oral function. Interestingly, however, while cGVHD oral pain and sensitivity is reported to interfere with eating, these symptoms are not significantly associated with weight loss and malnutrition, possibly because patients will either tolerate the pain and discomfort or modify their diet to minimize discomfort and maintain caloric intake . Patients with sclerotic involvement of the perioral tissues may report difficulty with oral function and hygiene and the ability to receive dental treatment .
Prevention and management of graft-versus-host disease
Systemic therapy for graft-versus-host disease
In general, the primary strategies to prevent/manage oral GVHD are centered on the strategies for the overall prevention and management of GVHD. Without prophylaxis, the incidence of aGVHD approaches 100%, and while GVHD prophylaxis protocols can reduce the risk, no specific prophylactic protocols have shown efficacy in preventing cGVHD . While it may initially seem desirable to entirely prevent GVHD, this potentially reduces the GVL effect and increases the risk of relapse-related mortality, whereas ineffective prophylaxis increases the incidence of GVHD with its associated significant morbidity and mortality.
Prevention of GVHD involves trying to control the immune reactivity of the grafted cells against host tissues . The primary approach for the prevention of GVHD is minimizing risk factors whenever possible; however, prophylactic regimens are necessary and are effective in reducing the incidence of aGVHD and include the use of low-dose methotrexate (MTX) alone or MTX plus steroids or a calcineurin inhibitor (cyclosporin or tacrolimum-506), with protocols using MTX with CSP being the most commonly used . The schedules for these regimens usually involve administration of MTX early post-alloHCT (eg, days 1, 3, 6, and 11 post-HCT) and CSP administration starting just before transplantation and extending as long as 6 months post-HCT. Additional studies are under way to determine the effectiveness of other combinations of immunosuppressive agents for the prevention of GVHD including FK-506, sirolimus, or mycophenolate mofetil (MMF).
If aGVHD is very mild (ie, grade I), prophylaxis will be continued and no additional treatment may be given, or possibly only topical treatments will be administered . For example, topical steroids can be used to manage mild localized aGVHD rashes. Oral beclomethasone is a highly potent steroid that is active locally for GI mucosal surfaces and has been shown to effectively manage GI aGVHD and thus reduce the need for systemic therapy . The use of psoralen plus ultraviolet A (PUVA) light has also been successfully used to treat “skin only” aGVHD.
The treatment of established Grade II or worse aGVHD varies from center to center and at times even between subsets of patients . High-dose systemic steroid therapy (2 mg/kg/d) is the mainstay of management, with a 20% to 70% response rate resulting in complete resolution without recurrence in 20% to 40% of patients . Calcineurin inhibitors, antithymocyte globulin (ATG), PUVA, or oral beclomethasone (possibly in combination with oral budesonide) will often be added in steroid-refractory cases , although the effectiveness of adding these agents to systemic steroid therapy has not been confirmed by large, randomized clinical trials. Novel approaches to manage steroid-resistant aGVHD are looking at the efficacy of combining steroids with newer agents such as sirolimus, anticytokine antibodies (anti–IL-2 receptor and etanercept, an anti–TNF-α antibody), pentostatin, denileukin diftitox, and monoclonal antibodies (visilizumab, alemtuzumab, infliximab, daclizumab, and ABX-CBL) .
The prevention of cGVHD primarily is hampered by an incomplete understanding of the pathophysiology of the disease. The most prominent risk factor for developing cGVHD has long been considered to be aGVHD, therefore prevention is organized around the same strategies used to prevent aGVHD, though the effect of these strategies is usually less clear or dramatic. Protocols have examined the effects of stem cell source and composition, graft manipulation, reduced intensity conditioning use of ATG, and duration of prophylactic immunosuppression on GVHD and have met with variable success . Pharmacologic approaches to prevent cGVHD have been not been encouraging.
Limited cGVHD, like limited aGVHD can often be managed locally with topical corticosteroids, topical tacrolimus, or PUVA . For instance, oral beclomethasone with or without budesonide can be very effective in managing limited GI GVHD. The initial treatment of extensive cGVHD depends on systemic corticosteroids (eg, prednisone 1.0 mg/kg/d) . Studies comparing prednisone alone or in combination with of CSP or FK-506 have generally been difficult to evaluate and have demonstrated variable benefit from the combined agent protocols, although this approach may reduce long-term complications of corticosteroid therapy . Patients at high-risk for cGVHD have shown modest benefit from combined steroid-CSP regimens, but for patients who had standard-risk CGVHD there was no benefit. Additionally, patients treated with prednisone plus CSP are at higher risk for fatal infections. The treatment of extensive cGVHD is subject to high failure and complication rates. Second-line therapies for steroid-refractory cGVHD include azathioprine (AZA), MMF, sirolimus, cyclophosphamide, thalidomide, rituximab, halofuginone and etretinate, daclizumab and etanercept, hydroxychloroquine, and extracorporeal photopheresis (ECP) .
Chronic GVHD tends to resolve slowly. Data from the Fred Hutchinson Cancer Research Center (FHCRC) has shown that for a cohort of 330 patients treated for cGVHD, approximately 33% required secondary systemic treatment in addition to steroids . Within the first 3 years during initial treatment, an additional 10% of the patients died from causes other than recurrent malignancy. Only 5% of patients were off of immunosuppressive therapy within the first year of diagnosis, and only 27% were able to discontinue primary treatment 3 years after diagnosis without the need for secondary treatment. Treatment results for steroid-resistant cGVHD are much less successful than situations where only steroid treatment is necessary. In a group of 104 FHCRC patients, therapies needed to be continued longer and with higher rates of nonrecurrent malignancy-associated mortality (19%), increased rate of development of second malignances (12%), and 13% of patients required continuation on treatment for steroid-resistant beyond 2 years; 48% required subsequent treatment with a variety of other immunosuppressive agents after HCT. It has been reported that 15% of HCT patients diagnosed with cGVHD were still on immunosuppressive therapy as long as 7 years after transplantation .
Management of oral graft-versus-host disease
Basic oral health
Maintaining oral and dental health and preventing oral infections is critical for patients with oral GVHD . Components of basic oral care include brushing, flossing, rinsing with bland agents (eg, normal saline), and xerostomia management with the primary goal being prevention of infections (eg, gingivitis, periodontitis, dental decay, and dental abscesses) ( Table 1 ).


