Abstract
Giant cell angiofibroma was first described as a distinctive orbital soft-tissue tumour in male adults; it is now recognized that this mesenchymal tumour can present in other anatomical regions. In this article, a case of giant cell angiofibroma of parapharyngeal space in a 25-year-old woman is described. Clinicopathologic features of this tumour are reviewed. To the authors’ knowledge, this is the first reported case of giant cell angiofibroma arising in the parapharyngeal space.
Giant cell angiofibroma (GCA) was described as a unique orbital neoplasm by Dei Tos in 1995 . This rare tumour is characterized by a patternless proliferation of spindle cells, multinucleated giant cells and pseudovascular spaces. Tumour cells stain positively for mesenchymal markers, CD34 and vimentin, and negative or minimal staining for epithelial, nerve and muscle markers. It is important to recognize this entity in order to avoid misdiagnosis with other fibrous and vascular tumours. The authors report the case of a GCA with a rare parapharyngeal location, and discuss its nature, diagnosis and treatment .
Case report
A 25-year-old woman was referred with a 1-month history of a left parotid tumour. Her medical history was unremarkable, except for the diagnosis of temporomandibular dysfunction syndrome. The patient had not undergone any previous surgery. There was no history of cancer in her family members. On examination, there was a large soft-tissue mass in the left parotid area. The painless lesion of fibroelastic consistency was palpated subcutaneously and intraorally along the pharyngeal area. Facial nerve function was intact and there was no evidence of palpable cervical lymphadenopathy. The remainder of the head and neck examination was normal ( Fig. 1 ).

A computed tomography (CT) scan performed with contrast revealed a well-delimited, encapsulated mass occupying the left parotid gland region in the deep lobe, that extended and obliterated the left parapharyngeal space and impinged on the left margin of the oropharynx. The mass had a well-circumscribed border with heterogeneous enhancement, measuring approximately 57 mm × 54 mm × 43 mm, without infiltration to the adjacent muscle or skin. No bone erosion was identified on imaging studies ( Fig. 2 ).

Fine-needle aspiration of the mass showed atypical myofibroblastic cells and intense haemorrhagic necrosis, consistent with a neoplastic process that could not be classified further. At this time, radiological and cytological differential diagnoses included parotid gland tumour, angiosarcoma, synovial sarcoma, fibrosarcoma and other pathological entities such as fibrous or vascular tumours.
Surgery was performed under general anaesthesia. The skin incision in the preauricular and cervical region allowed good access to the lesion for a conservative transparotid approach and safe control in case of a vascular accident. It was decided to perform an intraoperative histological examination of the parotid superficial lobe and two cervical lymph nodes. As there was no evidence of malignant cells in the lesion, the surgeon removed only the deep parotid lobe and parapharyngeal mass ( Fig. 3 A ). The tumour mass appeared to have no obvious infiltration into the surrounding tissues and was excised completely. The residual surgical defect was not reconstructed and the wound was drained and closed in layers.
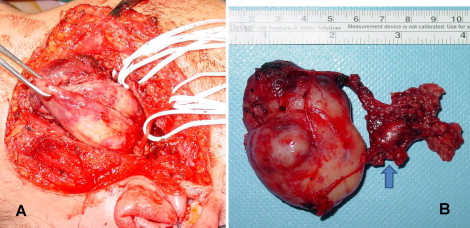
Macroscopic examination disclosed a solid, hard-elastic, brownish-red, lobular mass measuring 60 mm × 70 mm × 64 mm ( Fig. 3 B). Light microscopy examination revealed a circumscribed, cellular mass with a fibrous pseudocapsule. A proliferation of spindle cells containing round-to-oval bland nuclei with pseudoinclusions was present. Numerous floret-like multinucleated giant cells, capillary-sized blood vessels, and angiectoid pseudovascular spaces were present in the tumour ( Fig. 4 A and B ).

To rule out a diagnosis of synovial sarcoma, fluorescence in situ hybridization was employed with the LSI SYT Dual Color Break Apart rearrangement probe. A study of the SYT gene (18q 11.2) was made, with evaluation of at least 200 tumoural cell nuclei giving a negative result (rearrangement of the SYT gene in 8% of the tumour cells studied). Immunohistochemical analysis revealed strong positivity for the mesenchymal markers vimentin, CD34, BCL2 and CD99 in neoplastic cells. Tumour cells were negative for smooth muscle actin, CKAE1/AE3, CK1/5/10/14, S100, p63, CD31, EMA and PGFA. Ki67-positivity was evident in less than 10% of tumour cells. All of the immunohistochemical stains were compared with appropriate positive and negative controls ( Fig. 4 C and D).
The resected tissue was examined histologically by the pathology department and a diagnosis of GCA was made. The margins of the surgically resected tissue showed no evidence of tumour cells, and on that basis the authors concluded that the tumour had been removed successfully.
After surgery, the patient had an uneventful postoperative course with complete healing. She was discharged on the seventh postoperative day to be followed up at the outpatient clinic. At the 2-year follow-up examination she remained free of any recurrence of the condition.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


