DNA replication and gene expression
Publisher Summary
This chapter discusses Dna replication and gene expression. Research that led to the discovery that DNA is the genetic material began as early as 1928 and came from experiments with the Pneumococcus bacterium. This organism possesses a slimy polysaccharide capsule, which is required for pathogenicity in animals and enables it to form smooth colonies on agar plates. In 1928, Griffith found that if a mixture of live rough pneumococci and heat-killed smooth pneumococci were injected simultaneously into mice, they died from pneumonia. Further support for the genetic nature of DNA came later from studies with a virus, bacteriophage T2, that infects the bacterium Escherichia coli. Phage T2 is composed of a central DNA core surrounded by a protein coat. By differential labeling of the phage DNA with the radioactive isotope 32P, and the phage protein coat with 35S, Hershey and Chase were able to show that when T2 infects a bacterial cell, the phage DNA enters the host and is able to induce viral replication, whereas the phage protein coat remains outside the bacterial cell. The phage’s genetic information must reside within its DNA. DNA present in a double-stranded helical form is the genetic material in all prokaryotic and eukaryotic cells, although this is not always the case for viruses that may use single-stranded DNA, or even RNA, as their genetic material.
DNA–the genetic material
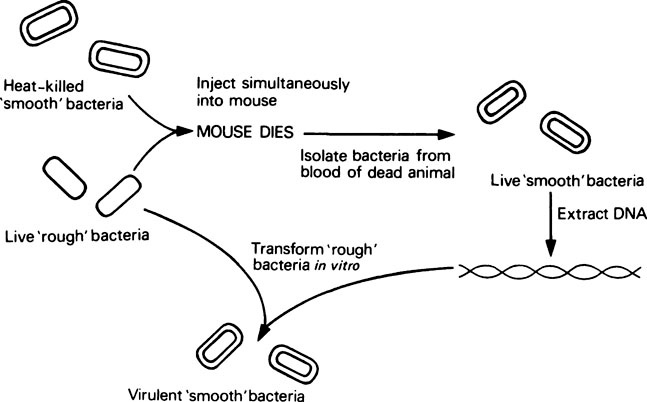
However, research which ultimately led to the discovery that DNA is the genetic material began as early as 1928, and came from experiments with the Pneumococcus bacterium. This organism possesses a slimy polysaccharide capsule which is required for pathogenicity in animals, and enables it to form ‘smooth’ colonies on agar plates. Mutants exist which cannot synthesize this capsule; they are therefore non-pathogenic and form ‘rough’ colonies on agar. In 1928, Griffith found that if a mixture of live ‘rough’ (non-pathogenic) pneumococci and heat-killed ‘smooth’ (pathogenic) pneumococci were injected simultaneously into mice, they died from pneumonia (Figure 20.1). Consequently a factor from the killed smooth cells was being transferred to the mutant rough cells, thereby making them pathogenic. This change or transformation was permanent and resulted in the appearance of live smooth pneumococci in the blood of the animal. Work then began on identifying the ‘transforming activity’ and in 1944 Avery and others finally showed that purified DNA extracted from killed smooth cells was able to transform unencapsulated rough pneumococci into encapsulated smooth cells in vitro, thereby establishing DNA as the transforming factor or genetic material.
The structure of genes
For any molecule to be recognized as the material responsible for transmitting genetic information, it must fulfil certain requirements dictated by our knowledge of the continuity of species and the process of evolutionary change. (1) It must contain biologically useful information in a stable form. This information is contained in the form of a linear array of genes, i.e. specific stretches of DNA, most of which code for a single protein. An average gene is perhaps 1000−2000 base pairs in length. (2) The genetic material must be capable of being accurately replicated, and transmitted faithfully from generation to generation. (3) The genetic material must be able to express itself in such a way as to direct the synthesis of all the different proteins characteristic of the organism, cell type or developmental stage. The amino acid sequence of each individual protein is determined by the nucleotide sequence of its corresponding gene(s). However, it will be seen later that proteins are not synthesized on the genes directly. Instead, an RNA intermediary, called messenger RNA (mRNA), is first synthesized (transcribed) using the DNA as a template. This mRNA then directs the synthesis of a specific polypeptide, the amino acid sequence being determined by the order of nucleotides in the mRNA which in turn reflects the DNA sequence from which it was transcribed. Gene expression therefore represents the flow of information from DNA to RNA to protein (page 202). (4) Finally, although in general the genetic information should be stable, it must nevertheless be capable of some variation or mutation, even if only very infrequently, to explain the observed characteristics of evolutionary change.
The bacterial chromosome
The whole of the genetic information contained within a single bacterial cell or eukaryotic nucleus is called the genome. In eukaryotes this is distributed between a number of different chromosomes, the number being characteristic of the species. Each eukaryotic chromosome is a highly condensed structure composed of both DNA and nuclear proteins, organized in a very specific manner (page 311), while the bacterial genome is present as a single chromosome (i.e. a single DNA duplex), with very little associated protein. The E. coli chromosome contains about 4 million base pairs in its DNA and is circular, although in vivo this circle must be folded very compactly since the contour length of the DNA is about 1000 times the diameter of the bacterial cell. A property of circular DNA molecules which contributes to this tight packing is supercoiling: the twisting of the double-helical axis itself to form a higher order of twisting, known as a superhelix or supercoil.
DNA replication
The Meselson−Stahl experiment
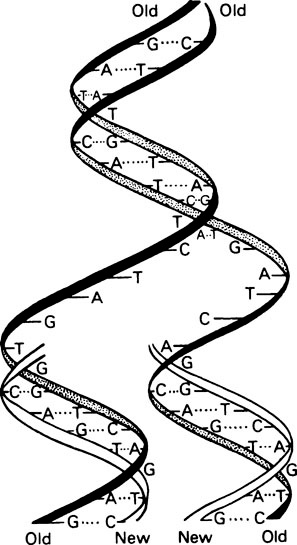
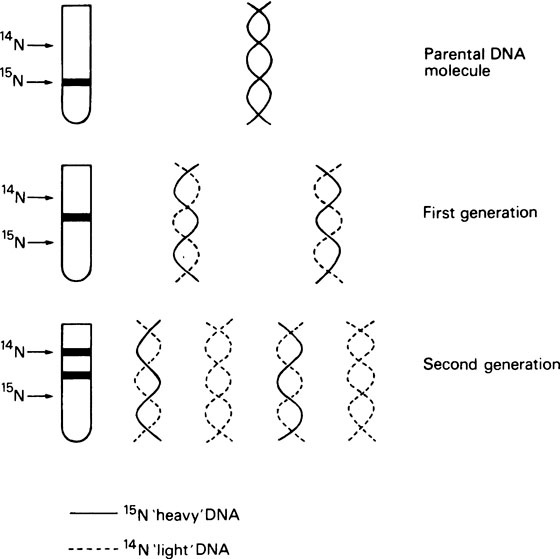
It is essential that DNA should be able to replicate since, after cell division, both daughter cells must each contain the genetic information. Watson and Crick suggested that this was possible with a double-stranded helix since the two strands could separate and each could then direct the synthesis of its complement by means of specific base-pairing (Figure 20.2). Such a mechanism would be described as semi-conservative since each daughter double helix would contain one parental DNA strand and one newly synthesized strand. Semi-conservative DNA replication was actually shown to occur by Meselson and Stahl. For this they labelled E. coli DNA with a ‘heavy’ isotope of nitrogen, 15N, by growing the bacteria for many generations in 15N-labelled ammonium chloride as the sole nitrogen source. Some of the bacteria were removed for analysis immediately and the remainder were transferred to a medium containing normal 14N ammonium chloride. Cells were removed from this medium at intervals and their DNA extracted, and analysed by isopycnic or equilibrium density gradient centrifugation. This technique involves placing a tube of concentrated caesium chloride (CsCl) solution in an analytical ultracentrifuge. The CsCl, because it is denser than water, tends to sediment to the bottom of the tube when centrifuged at high speed. However, the tendency to sediment is opposed by diffusion and after a number of hours an equilibrium is reached with the CsCl forming a continuous stable density gradient. If a mixture of macromolecules is dissolved in the original CsCl solution they will come to equilibrium at a position in the gradient where the density of the CsCl solution exactly matches their own, causing them to separate into bands, the positions of which can be located by optical means. This technique is sufficiently sensitive to allow the separation of ‘heavy’ DNA (containing 15N) from normal ‘light’ DNA (containing 14N). Their results are summarized in Figure 20.3, which shows that after one bacterial generation, centrifugation of purified DNA produced a single band with a buoyant density intermediate between that of the original 15N-labelled ‘heavy’ DNA and ‘light’ DNA from cells grown only on non-isotopic medium. After two cell generations, two DNA bands of equal intensity were obtained; one of hybrid density, and one of ‘light’ (14N) density. These were exactly the results predicted for semi-conservative replication.
The mechanism of DNA synthesis
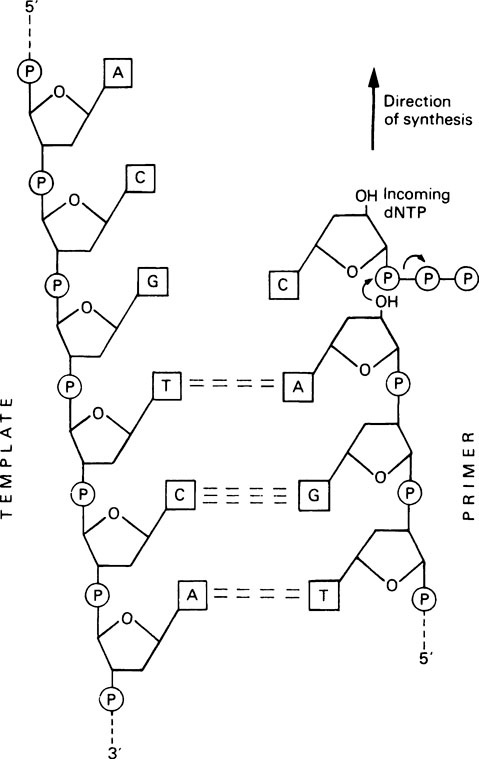
DNA synthesis proceeds by a nucleophilic attack by the 3’-OH group of the primer on the α-phosphorus atom of the incoming deoxynucleoside triphosphate, to form a phosphodiester bond (Figure 20.4). Pyrophosphate is released and is hydrolysed by pyrophosphatase, thereby driving the polymerization reaction in the forward direction. Synthesis therefore occurs in a 5′ to 3′ direction; the enzyme remains attached to the DNA throughout and is not released after each nucleotide addition.
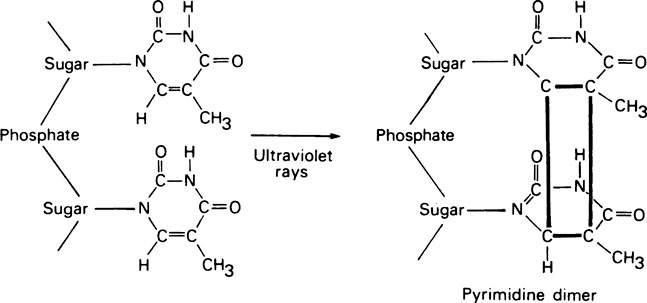
DNA polymerase I also has another exonuclease activity, but in this case hydrolysis occurs in a 5′ → 3′ direction and appears to serve two functions in vivo; (i) it is involved in removing RNA primers during DNA replication (see later) and (ii) it plays a role in DNA repair processes. That this is an important function is illustrated by experiments in which E. coli was exposed to ultraviolet light, which in large doses can cause adjacent pyrimidine residues situated in the same polynucleotide strand to become covalently cross-linked (Figure 20.5). Whereas normal E. coli cells are able to excise these pyrimidine dimers and repair their DNA, a mutant form which lacked DNA polymerase I was unable to do so and was therefore more readily killed by ultraviolet light. In humans, as in E. coli, exposure to ultraviolet light causes the production of stable pyrimidine dimers but, as shown in studies on skin fibroblasts, these are rapidly excised from the DNA of normal subjects. Sufferers from the rare inherited condition of xeroderma pigmentosa, however, appear to lack the enzyme responsible for nicking the DNA backbone in the vicinity of the pyrimidine dimers and skin cancer is a frequent result.
DNA replication in bacterial cells
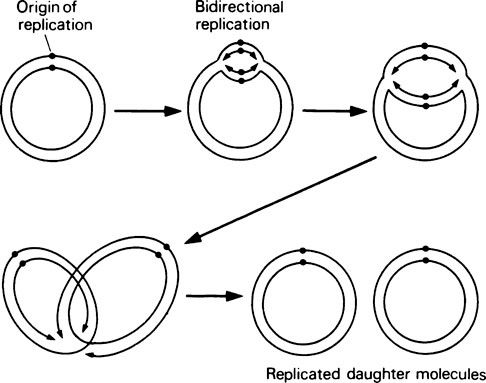
DNA replication in E. coli (Figure 20.6) has the following properties. (a) Replication starts at a unique site on the circular bacterial genome, called the origin of replication. (b) Replication proceeds simultaneously in both directions around the genome, that is, it is bidirectional. In other words, there are two replication forks, one travelling clockwise and one anticlockwise, at about the same rate. (c) Replication is terminated at a point diametrically opposite the origin of replication.
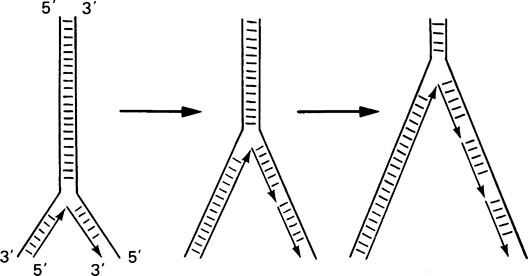
Such an overall mechanism poses a number of problems at the molecular level. First, at a replication fork, both strands of the double helix must be replicated, but since they are antiparallel this would suggest 5′ → 3′ synthesis on one strand and 3′ → 5′ on the other; yet DNA polymerase can only synthesize in the 5′ → 3′ direction. This dilemma was resolved by Okazaki who found that one DNA strand is synthesized continuously in the expected 5′ → 3′ direction, whereas the opposite strand is synthesized discontinuously as short pieces of DNA known as Okazaki fragments (Figure 20.7). Each of these fragments is synthesized in the expected 5′ → 3′ direction and adjacent pieces are rapidly joined together by another enzyme called DNA ligase giving the appearance that overall growth is in the 3′ → 5′ direction.
The second problem is that the initiation of DNA synthesis requires a primer, base paired to the template, and each Okazaki fragment requires its own primer. In fact there is a specific RNA polymerase, called primase, which can synthesize short RNA primers of about ten nucleotides. RNA synthesis, unlike DNA synthesis, does not require a primer for initiation (page 000). These short RNA molecules then act as primers for DNA polymerase (Figure 20.8). It is now known that DNA polymerase I is not the major replicating enzyme in E. coli. Instead another enzyme, DNA polymerase III, carries out most of the replication in vivo, whereas DNA polymerase I is responsible for degrading the RNA primers after DNA synthesis by means of its 5′ → 3′ exonuclease activity and replacing it with DNA using its polymerase and 3′ → 5′ proof-reading activities.
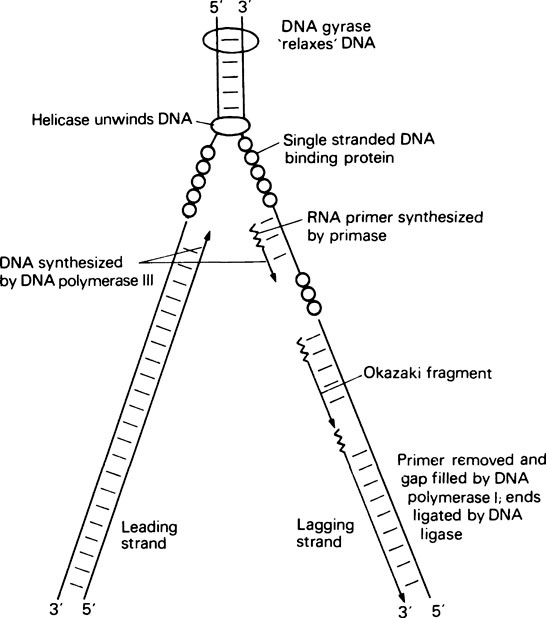
The final problem in DNA replication is the unwinding of the double helix that is necessary for replication to occur, and the subsequent rewinding of the daughter molecules. The whole molecule does not unwind. Instead a small region of the helix around the replication fork is unwound with the aid of a number of specific proteins (Figure 20.8) including a helicase, which carries out the actual unwinding, and single-stranded DNA binding protein, which stabilizes the single strands and prevents them from base pairing. Finally, when the DNA helix of a circular genome is unwound this puts a tortional strain on the rest of the molecule. This strain is relieved by DNA gyrase which acts as a ‘molecular swivel’ and allows the DNA to relax. Having successfully unwound a region of double-stranded DNA, primase can then bind and synthesize an RNA primer, DNA polymerase III can bring about DNA synthesis, DNA polymerase I can replace RNA primer with DNA, and finally DNA ligase can join adjacent Okazaki fragments.
Gene expression − transcription
As already stated DNA is not itself the direct template which determines the order of amino acids during protein synthesis, since protein synthesis can be carried out in vitro in the absence of DNA. Furthermore, protein synthesis in eukaryotic cells occurs in the cytoplasm whereas DNA is confined to the nucleus (and mitochondria; page 201). Instead, a particular class of RNA molecule, the messenger RNAs (mRNA), act as templates for protein synthesis. Each mRNA has a nucleotide sequence which is complementary to that of the gene from which it is transcribed. Hence the mRNA is capable of transmitting the genetic information contained within that gene. The existence of mRNA was first shown during experiments carried out on E. coli cells infected with bacteriophage T2. Following infection, nearly all the proteins synthesized by the host cell are phage-specific not host-specific proteins, i.e. they are genetically determined by the phage DNA. A minor RNA fraction, with a very short half-life, was found to appear soon after infection. This RNA had a base composition which was complementary to that of one of the phage DNA strands but was unlike that of E. coli
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


