Chapter 29
Cleft Lip and Palate: An Overview
Global Burden of Birth Defects: Cleft Lip and Palate
More than four million children are born with birth defects worldwide every year. Craniofacial anomalies comprise a large fraction of all human birth defects, less frequent only than congenital heart disorders and clubfoot. Cleft lip with or without palate (CL/P) is the most common craniofacial birth defect with an estimated quarter of a million affected babies born each year in the world. This malformation shows considerable variation across geographic regions and ethnic groups and has significant medical, psychological, social, and economic ramifications. It is a costly public health problem with an average lifetime treatment cost per child in the US estimated to be roughly $101 000.
The World Health Organization (WHO) and most cleft organizations across the globe recommend interdisciplinary care by a team of specialists. In reality, however, surgical and non-surgical treatment is often fragmented and dictated by socio-economic factors and access to medical facilities. In developing countries, particularly in rural areas, care is often neglected due to social beliefs and lack of awareness, or initiated late due to restricted resources and inadequate access. The delay in treatment and intermittent care by local or overseas cleft mission surgeons, combined with incomplete follow-up, results in poor outcomes with unnecessary complications. More recently, some humanitarian non-profit organizations supporting cleft care have changed their aid philosophy. They are identifying centers with a potential to deliver quality care in low- and middle-income countries and providing support for the physicians, staff, and hospital infrastructure, helping to establish parameters of care, as well as monitoring treatment outcomes at these centers.
Birth defects are emerging as a cause of neonatal mortality in countries that have made progress in controlling infectious diseases and malnutrition. The strategies proposed to reduce the global impact of birth defects include: (1) effective family planning, genetic counseling, and prenatal diagnosis; (2) education for couples to decrease maternal exposure to avoidable environmental risk factors such as tobacco, alcohol, and teratogenic medications; (3) improving periconception maternal intake of micronutrients such as folic acid (400 μg); and (4) improving the availability of medical and surgical care locally for the affected infants. National leadership and commitment are essential for proper surveillance of birth defects, infant mortality, and to monitor the clinical and cost effectiveness of various interventions.
The WHO initiative for collaborative craniofacial anomalies research has identified areas of uncertainty in clinical care and efforts are being made to conduct trials with sufficiently large samples of patients to provide evidence for treatment strategies. Initial research efforts have focused on addressing surgical, anesthetic, and nursing care for patients with craniofacial anomalies in developing countries. Surgical techniques for repair of various cleft sub-types and correction of velopharyngeal insufficiency are being evaluated. Adjunctive services such as use of prophylactic ventilation tubes, presurgical orthopedics, psychological counseling, speech therapy, and feeding interventions before and after surgery are also being monitored and assessed. An international database of craniofacial anomalies has been established to improve answers to questions relevant to individuals with cleft and craniofacial anomalies, their families, and health care providers.
Epidemiology
Cleft lip with or without palate (CL/P) has an average birth prevalence of 1 : 700 ranging from 1 : 500 to 1 : 2000, depending on the race (Table 29.1). There are wide ethnic variations with highest occurrence in Native Americans (3.6 : 1000), followed by Asians (2.1 : 1000 Japanese births and 1.7 : 1000 Chinese births), Caucasians (1 : 1000), and lowest in those of African descent (0.3 : 1000). Cleft of palate only (CP), which differs genetically from CL/P, has a birth prevalence rate of 1 : 2000 and is more similar across all populations. About half of the oral clefts involve lip and palate (46%), a third of the clefts involve only the palate (33%), and clefts of lip alone account for 21%. CL/P is more often unilateral than bilateral and more common in males than females. The unilateral defects occur more often on the left side than the right side. Clefts of lip occur in the ratio of 6 : 3 : 1 for unilateral left, unilateral right, and bilateral. CP is more common in females and more often associated with other developmental anomalies.
Table 29.1 Epidemiology of oral clefts.
| Distribution of oral clefts |
| Cleft lip and palate 46% |
| Cleft palate only 33% |
| Cleft lip only 21% |
| Cleft lip with or without palate |
| Average birth prevalence 1 : 700 |
| More common in males |
| Unilateral > bilateral |
| Left side > right side |
| Association with other anomalies 10% |
| Cleft palate only |
| Average birth prevalence 1 : 2000 |
| More common in females |
| Association with other anomalies 50–60% |
Clefts are referred to as non-syndromic and syndromic, based on their association with other anomalies. About 50% of CP and 10% of CL/P are associated with a syndrome. Some common syndromes associated with cleft lip and palate include Van der Woude, Treacher Collins syndrome, Down syndrome, oro-facial digital syndrome, Opitz syndrome, craniofacial microsomia, and fetal alcohol syndrome. Nearly half of the syndromic cleft palate presentations are associated with the triad of micrognathia, glossoptosis, and airway obstruction (Pierre Robin sequence). The most common syndromic presentations of this triad are Stickler’s syndrome, accounting for 25%, and velo-cardio-facial (VCF) syndrome, accounting for 15% of all syndromic cleft palate individuals.
Etiology and Genetics
Non-syndromic CL/P is a complex trait with multifactorial etiology, resulting from gene–gene and gene–environmental interactions. Identification of key genes contributing to the genesis of orofacial clefts will help in early diagnosis, disease prevention, or possibly developing adjunctive therapies. The most recent estimates suggest that anywhere from 3 to 14 genes contribute to cleft lip and palate. Candidate genes and loci responsible for non-syndromic CL/P have been identified on chromosomes 1, 2, 4, 6, 11, 14, 17, and 19. Two genes IRF6 and MSX-1 now seem to explain about 15% of non-syndromic CL/P. Mutations in IRF6 lead to Van der Woude and popliteal pterygium syndromes. Mutations in other genes, TBX22, FGFR1, and P63, also contribute to syndromic clefts. Aberrant transforming growth factor beta-3 (TGF-β3) signaling plays a role in the pathogenesis of cleft palate.
Environmental factors that contribute to the etiology of facial clefting disorders include cigarette smoking, folic acid deficiency during the periconceptional period, and maternal exposure to alcohol and teratogenic medications such as retinoids, corticosteroids, and anticonvulsants (phenytoin and valproic acid). Co-sanguinous marriages, maternal diabetes, and obesity have also been linked to an increased risk of orofacial clefts. Less consistent associations have been found between clefts and maternal viral infections such as rubella and varicella.
Studies conducted to determine the risk of having a child with CL/P show that every parent has about a 0.14% (1 : 700) chance of having a child with a cleft. The risk of recurrence of a cleft condition is determined by a number of factors, including the number of family members with clefts, their relationship to family members with clefts, race and sex of the affected individuals, and the type of cleft. Studies show that the recurrence risk for first-degree relatives is about 3.3% for CL/P and for isolated CP it is 2%. Once parents have a child with a cleft the risk of having a second child with a cleft is about 2–5%, and after two affected children that risk rises to 9–12%. In twins with CL/P and those with isolated CP, the concordance is far greater for monozygotic twins than for dizygotic twins. Parents and young adults should be counseled appropriately by a geneticist so that they are in a better position to make decisions about future pregnancies.
Embryology
The embryologic development of the face begins at 4 weeks after conception from the neural crest ectomesenchyme that forms five prominences; the frontonasal process, and paired maxillary and mandibular processes surrounding a central depression. During the fifth and sixth weeks of embryonic development, bilateral maxillary processes derived from first brachial arch fuse with the medial nasal process to form the upper lip, alveolus, and the primary palate (Fig. 29.1). The lateral nasal process forms the alar structures of the nose. The lower lip and jaw are formed by the mandibular processes.
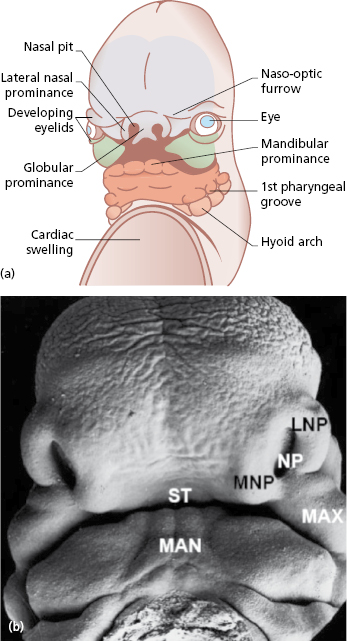
This process of formation of the face is the consequence of a cascade of processes that involve cell proliferation, cell differentiation, cell adhesion, and apoptosis. Failure or error in any of these cellular processes that lead to fusion of the medial nasal process with the lateral nasal and maxillary process can cause orofacial clefts (Fig. 29.2). The molecular events that underlie these cellular processes are under the control of a strict array of genes that include fibroblast growth factors (FGFs), sonic hedgehog (SHH), bone morphogenic proteins (BMPs), and members of the transforming growth factor beta (TGF-β) superfamily and other transcription factors. The formation of the secondary palate begins during the sixth week after conception from the two palatal shelves, which extend from the internal aspect of the maxillary processes. During the eighth week, these bilateral maxillary palatal shelves after ascending to an appropriate position above the tongue, fuse with each other and the primary palate. A disruption in the fusion of these embryonic components can occur due to delay in elevation of the palatal shelves from vertical to horizontal, defective shelf fusion, or post-fusion rupture resulting in a cleft of the secondary palate (Fig. 29.3).
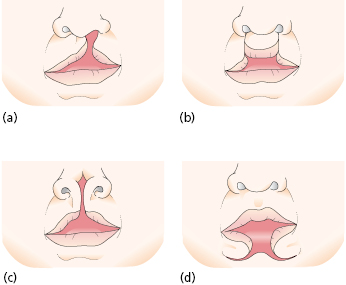
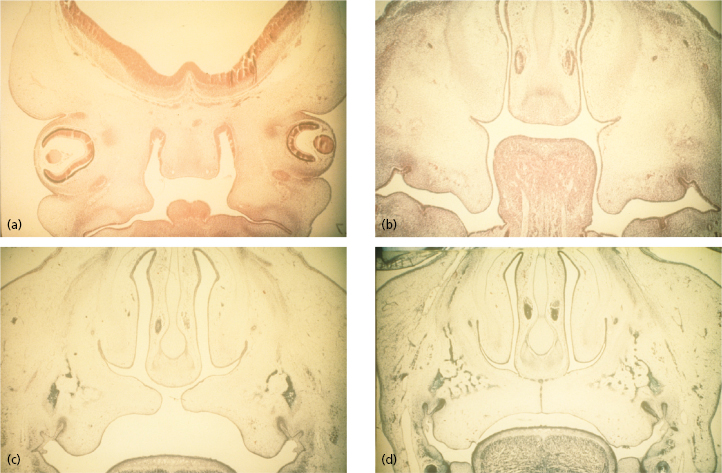
Classification
In order to standardize documentation and communicate effectively, various types of classification systems have been described. The early Veau classification included groups 1–4 with increasing severity of clefting:
- group 1 – cleft of the soft palate;
- group 2 – cleft of the hard and soft palate up to incisive foramen;
- group 3 – complete unilateral cleft lip and palate;
- group 4 – complete bilateral cleft lip and palate.
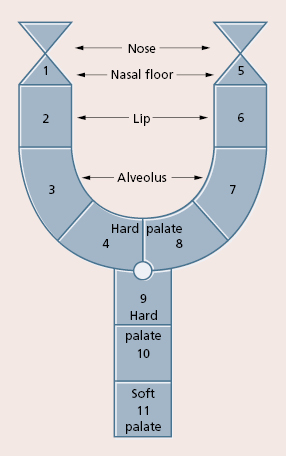
However, this classification is not always adequate to document the variations. The more sophisticated schematic diagrams, such as the one described by Kernahan and Stark have been used recently (Fig. 29.4). Berkowitz used a simple classification for labio-palatine clefts:
- Clefts of lip and alveolus.
- Clefts of primary (including lip) and secondary palate.
- Clefts of secondary palate only.
- Submucous cleft.
Interdisciplinary Management of the Cleft Individual
The idea of interdisciplinary care is to coordinate treatment by multiple specialists in a timely fashion with an aim of achieving normality in all aspects, including feeding, breathing, speech, hearing, alignment of teeth, appearance, and overall psychological and physical development. The timing of surgical and non-surgical interventions should coincide with the physical, cognitive, and social development of the child (Table 29.2). Cleft teams generally include a craniomaxillofacial surgeon, pediatrician, nurse practitioner, speech pathologist, orthodontist, social worker, and geneticist. Experience in Scandinavian countries and a multicenter Euro-cleft study have demonstrated that standardization, centralization, and participation of surgeons who perform a large number of cleft procedures produce better surgical results in terms of speech, appearance, and facial growth. However, this cannot be applied to all countries, particularly those with a high volume of cleft individuals and a limited number of care facilities.
Table 29.2 Timing of treatment in the cleft patient.
| Prenatal |
| Diagnosis and parental counseling |
| 0–6 months |
| General assessment for associated anomalies |
| ENT evaluation – breathing, feeding, swallowing, and hearing |
| Presurgical orthopedics (0–3 months) |
| Primary lip repair (3–4 months) |
| 6 months–2 years |
| Speech and oral sensory motor assessment |
| Grommets/ear tubes (as needed) Primary palate repair (9–12 months) |
| Preschool: 3–5 years |
| Dental care |
| Speech assessment and therapy (continue as needed) |
| Assess need for lip revision |
| Childhood: 6–12 years |
| Correction of velo-pharyngeal dysfunction (as needed) |
| Orthodontic treatment – phase I |
| Alveolar cleft repair (8–11 years) |
| Adolescence: 13–18 years |
| Orthodontic treatment – phase II |
| Orthognathic surgery (if needed) – 14–16 years (female), 16–18 years (male) Revision chielorhinoplasty |
| Replacement of missing teeth (as needed) |
Prenatal Diagnosis
Interdisciplinary team care begins with prenatal diagnosis and parental counseling. With the advent of sophisticated high resolution three-dimensional (3D) ultrasonography and genetic tests for screening of birth defects, intrauterine diagnosis of cleft lip is possible. Early diagnosis of a cleft of the lip should alert the obstetrician to the possibility of other malformations that may require further investigations. While early diagnosis may help parents to be better prepared, the advent of such a capability raises both ethical and psychological issues, such as dilemma of termination of birth. Physicians and surgeons have to inform parents that CLP in the absence of other major systemic anomalies is a treatable non-life-threatening condition. The cleft team also can discuss feeding issues and timing of lip and palate surgery, and can help establish contact with support groups for the family.
Transvaginal ultrasonography may reveal a cleft of lip as early as 11 weeks whereas 16–20 weeks is ideal for transabdominal ultrasonography. Several factors may influence the accuracy of ultrasound studies: sophistication of the scanning equipment; experience and skill of the sonographer; number of weeks into pregnancy; position of the baby while scanning; amount of amniotic fluid; maternal body structure; and severity of cleft.
Premaxillary protrusion is an important clue to the presence of cleft lip and cleft palate and may be more conspicuous than the cleft itself. The presence of a paranasal echogenic mass favors the presence of bilateral cleft lip and cleft palate. Clefts of palate alone are rarely visualized on ultrasound. The majority of the cases of orofacial clefts are detected antenatally. A recent study by Johnson et al. showed that the frequencies of prenatal diagnosis for cleft lip and palate, cleft lip only, and CP only were 33.3%, 20.3%, and 0.3%, respectively. Although the benefits of fetal healing have been well documented, at this time there is no indication for intrauterine repair of the cleft lip deformity as the risk of fetal surgery is far too high both for the fetus and mother for correction of this non-life-threatening condition.
General Assessment
Every child born with a CL/P should be thoroughly assessed by complete physical examination and necessary diagnostic tests to check for associated systemic abnormalities, including congenital heart, renal, or airway anomalies. If the child is delivered in a non-medical facility or a small hospital they should be referred to a tertiary hospital with specialists or a craniofacial team for further evaluation. A proper airway assessment, and counseling for nutrition and feeding should be initiated immediately.
Feeding and Nutrition
Feeding is one of the first concerns in a child born with a CL/P. Parents should be taught how to feed the baby and informed about various feeding nipples that can deliver more milk under less pressure. The goal is to provide adequate nutrition to satisfy the caloric requirements and avoid failure to thrive. The team nurse generally provides the parents with information on feeding before birth or immediately after birth. Children with cleft lip only without a cleft palate may have some difficulty in creating a seal around the nipple but generally can be breast fed before and after lip surgery. However, the presence of a cleft palate makes it difficult to create a negative pressure that is necessary to feed. Four randomized clinical trials conducted with a total of 232 babies showed that squeezable bottles may be easier to use than rigid bottles for children with CL/P. There was no statistical difference found in growth outcomes of children with CL/P who were fitted with a passive palatal appliance to help with feeding and those without an appliance. A feeding tube is rarely necessary except in infants with other associated anomalies, particularly airway anomalies.
Ear, Nose, and Throat Evaluation
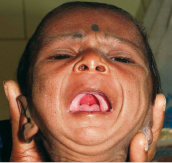
A proper airway assessment should be priority for a newborn with congenital craniofacial anomalies. Infants are obligate nasal breathers. It is important to check if there is obstruction at any level in the upper or lower airway, including the nares and choanae. Children born with a cleft of the palate may have associated micrognathia, glossoptosis, and airway obstruction (Fig. 29.5). In these children, one should look for signs of increased effort while breathing, stridor, weight loss, and failure to thrive. Parents should be informed to watch for an abnormal breathing pattern or respiratory distress, particularly during upper respiratory tract infection. If there are signs of airway obstruction, a pediatric otolaryngologist should be consulted to perform an endoscopic evaluation of upper and lower airway to look for possible cause of obstruction.
An audiology assessment is recommended soon after birth to check for hearing abnormalities. Children with cleft palate exhibit a higher frequency of otitis media prior to palate repair than those without clefts. Middle ear ventilation disorders due to Eustachian tube dysfunction can cause conductive hearing loss. This can also contribute to speech and language delay in these children. Although not as common as conductive hearing loss, sensorineural hearing deficits exist within the cleft population, and it has an effect on speech perception and clarity, as well as auditory compr/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


