Corrosion
Corrosion is the chemical reaction of a metal with components of its environment. Since in the dental or more general biomaterials context metals may be exposed to wet warm, salty, acidic oxygenated conditions the possibility of such reactions must be considered. This chapter sets out the types of corrosion mechanism, methods of control, factors influencing the outcome. This is to enable the correct decisions to be taken in choosing alloys for specific applications the recognition of risk factors to achieve the most favourable long-term solution in treatment.
Corrosion is an electrochemical process, and the electrode processes operating in spontaneous and driven corrosion systems are explained. The identification of what types of reaction and where they are occurring are key issues.
There are a number of methods of protection against corrosion which are in common use, whether by providing a physical barrier or deliberate control of the corrosion reaction, but unfortunately very few possibilities are appropriate or feasible in the oral environment. In addition, corrosion once started tends to be self–perpetuating.
Two approaches are feasible: the use of either inert or passive metals or alloys. Passivity is obtained through an unreactive oxide coating, but this is not a guaranteed cure. Corrosion can still occur under strongly acidic or alkaline conditions, and then can continue in a much more severe fashion. Even so, passive metals are of increasing importance in dentistry, especially titanium.
Stress corrosion is a risk whenever a metal object is stressed under potentially corrosive conditions as the stress increases the driving force for the reaction. This applies whether the stress is continuous or intermittent.
Electrochemical processes are also used for deliberately etching or polishing metals as well as for plating. These techniques allow a close control that would otherwise be difficult to achieve, and in some cases permit a process that would not be practical another way.
A major factor in the design and selection of alloys for use in dentistry is the corrosion resistance. It is only by being aware of the factors involved, and understanding the mechanisms and processes operating, that the correct choices can be made for effective long term treatment. There are also implications for the tools and instruments used in dentistry, where sterilization offers more serious challenges.
There are a number of applications for metallic materials in dentistry, both within the mouth and in the numerous instruments, tools and equipment associated with clinical and laboratory work. The prime demands on a metallic structure are usually those of strength and rigidity, but it seems self-evident that in addition there must be a lack of chemical reaction with the substances found in the working environment. It is plain that not all metals are as unreactive as gold or platinum, and the chemistry of a metal or alloy must therefore be taken into account when designing for a particular application. The more aggressive the environment, the more serious the problem. This aspect of metal chemistry is usually referred to as the corrosion properties; corrosion resistance or corrosion rate are the relevant concerns.1 In particular, the oral environment presents a corrosion challenge to metallic devices: it is warm, wet, acid and salty. While these conditions are physiologically benign and normal (and not obviously challenging), for many metals they represent substantial problems, especially in the context of the many years of exposure that are expected. To understand, therefore, the design of alloys for dental use, and the limitations in application or handling that arise from their corrosion behaviour, the principles must first be established.
The corrosion of metallic objects has a number of possible consequences in the dental or biomedical context. Firstly, the metals most often encountered as structural materials are those from the transition periods of the table of the elements, groups VIA – IIB. Their oxides and salts are typically strongly coloured (24§6). Thus, their corrosion products will tend noticeably to discolour the metal itself, if they adhere, or the surrounding tissue or other materials. Secondly, the fact that metal is being removed from the surface of the object by the corrosion reactions means that its roughness may increase. This would spoil the appearance if the object was originally highly polished but also, in the mouth, it would be more retentive of plaque, in itself undesirable. The loss of material may go further, perhaps intergranularly or in pitting, and reduce the mechanical strength of the object, causing failure. Lastly, except for a few that are required in very small amounts for special physiological or biochemical reasons (the so-called ‘trace’ elements) the ‘heavy’ metals are mostly toxic; iron is the obvious exception to this. Corrosion products therefore may pose a threat of local or systemic effect on the organism. These problems are not mutually exclusive, and various combinations usually occur. Whatever the combination, from the point of view of the task being performed by the metal object, corrosion of any kind is usually undesirable. It must therefore be avoided or controlled to be very limited.
§1 Basic Considerations
•1.1 One electrode
Whenever a metal is in contact with an aqueous solution such as saliva or blood (Fig. 1.1) there is a spontaneous tendency for metal ions to go into solution, leaving electrons behind. This reaction may be written:
< ?xml:namespace prefix = "mml" ns = "http://www.w3.org/1998/Math/MathML" />
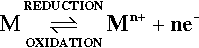
Notice that the reaction moving to the right involves the removal of electrons, the oxidation of metal atoms to positive ions (cations), while the converse reaction is a reduction of the cations to metal, i.e., the addition of electrons.
Because a charge separation is involved in the process of oxidation (i.e. work is being done), it is appropriate to define thetendency of the reaction to move to the right by a voltage or potential difference between the solution and the metal immersed in it. This might be termed the escaping tendency for the ion. There is no way to measure this voltage directly because any other contact with the solution would involve a similar reaction, operating in the opposite direction, obscuring the value of interest. Even so, it can be readily appreciated that one factor of relevance in establishing the equilibrium must be the effective concentration or activity of the metal ion in the solution, and so the electrode potential is dependent on the composition of the solution. The potential is also dependent on temperature (equation 8§3.1). A metal in contact with a solution in this manner is termed an electrode.
•1.2 Galvanic couple
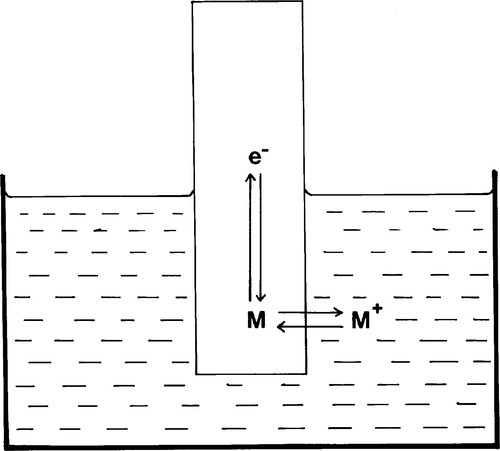
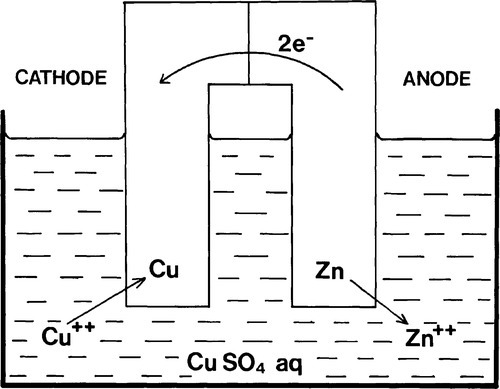
If two different metals are immersed in the same solution (without touching), the equilibria set up will be largely independent of each other. But if the metals, say Cu and Zn, are in contact (Fig. 1.2), allowing electrons to move freely between the two, and the escaping tendencies of their ions are different, only one reaction can move to the right. Essentially this is because the higher electron ‘concentration’ resulting from metal with the higher tendency to dissolve unbalances the equilibrium (1.1) from the point of view of the second metal, forcing reduction. Thus, should a supply of the other metal’s ions be already available from the solution, the reaction in respect of that metal will move to the left, and metal atoms will be deposited on that electrode.
The electrode associated with the metal ion reduction process is known as the cathode, and that associated with the oxidation process is the anode. This pair of definitions represent the single most important distinction to remember for, once having identified an electrode process, all else follows.
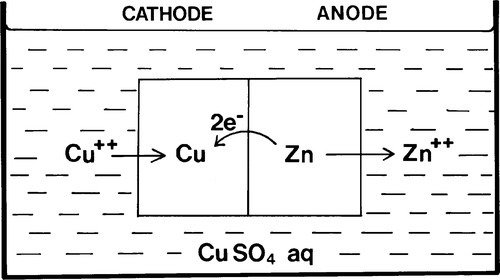
Thus, in the course of this spontaneous reaction, summarized as:
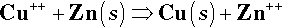
(where the ‘s’ refers to the solid state), electrons must be transferred from one electrode to the other. It does not matter what path is taken so long as the electrical connection is made, and this may easily be outside of the solution or electrolyte (Fig. 1.3). In comparison with the arrangement of Fig. 1.2 there is no change in any aspect of this system electrochemically, assuming that there is no electrical resistance in the external circuit.
If such a resistance is incorporated, however (Fig. 1.4), the transfer of electrons from one side to the other is delayed, creating a backlog as it were. The net surplus of electrons at their source (the anode) leads to this electrode carrying a negative charge, while the deficit at the cathode leads to it carrying a positive charge. The voltage measured now across the terminals, if the resistance is very large, corresponds to the potential difference between the electrodes. Such a system of dissimilar metals with an electrical connection, both immersed in an electrolyte, is known as a corrosion cell or galvanic couple.2 The only requirement for the relevant reactions to tend to go is that the electrode potentials of the two metals, under the prevailing conditions, are different.
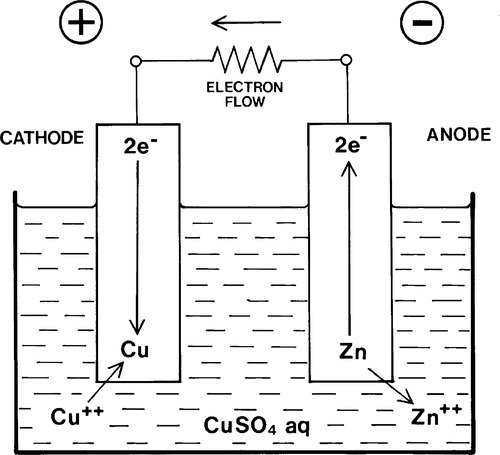
Note that it is incorrect to say that the potential difference measures the rate of reaction. The rate also depends on circuit resistance and concentrations, as well as other factors.
•1.3 Chemical potential
If we write a hypothetical equilibrium constant equation for the anode reaction (1.1):
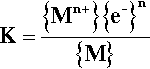
not only does K depend on the metal ion activity in the solution(as already discussed) and on the ‘electronactivity’ or potential at that point, there is also a term for the activity of the solid metal (8§3.2). This is normally by convention taken to be unity, which can be interpreted very simply as meaning that the activity of the solid is independent of its bulk – remembering that thermodynamics does not consider the quantity of material as such. However, this assumes that the metal is in a standard state, usually understood to be a perfect, perfectly pure crystal. The actual activity is affected by lattice defects, strain, roughness, temperature, impurities and deliberate alloying, all of which affect the energy of the system.
If the atoms of the metal of interest are not in pure solid but dissolved in another metal, its chemical potential is evidently altered (cf. 8§3.2), and so will be its electrode potential too, and in a concentration-dependent manner (albeit not necessarily in a linear fashion). So the electrode potential of a single-phase alloy will reflect the elements present as well as their proportions. The electrodes of the system shown in Fig. 1.4 could just as well be of Cu-Sn and Sn-Zn alloys3 as of the pure metals. The potentials will be different and the rate of reaction may be different, but the net effect will be the same. Even single-phase alloys from the same two metals but with different compositions, for example α and β Ag-Cu (Fig. 12§3.1), will show this kind of electrochemical behaviour because each component may have different potentials, both electrical and chemical.
•1.4 Driving forward
The rate of the electrode reactions in Fig. 1.4 depends on the diffusion of Cu ions to the cathode, the diffusion of Zn ions away from the anode (to permit the reaction 1.1 to move to the right), and transfer of electrons from anode to cathode. Stirring would obviously help the first two, but the rate could also be increased if the electrons could be delivered to the cathode at a higher rate. So if some kind of electron ‘pump’ were available to charge up the copper electrode with more electrons so that it acquired a lower positive charge or even a net negative charge (Fig. 1.5) when immersed in the electrolyte, the same reactions would still go but at a greater rate (Fig. 1.6).
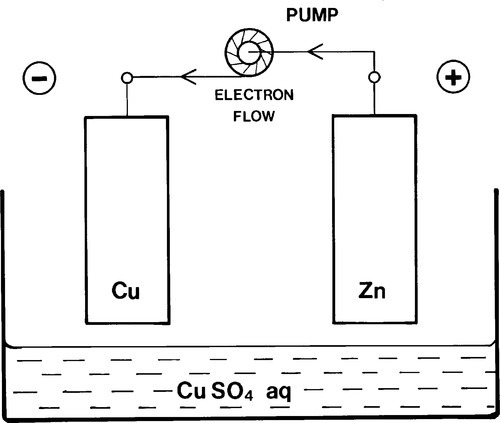
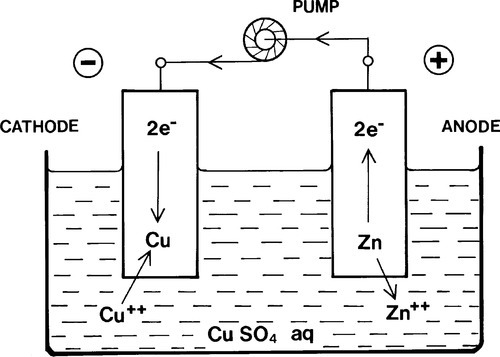
Notice that the charges on the electrodes are now of opposite sign to those in Fig. 1.4, although electrochemically there is no change in the description. In other words, the charge on an electrode is no guide to the electrode process occurring at its surface. This illustrates the fundamental point stressed above on the nature of electrochemical cells, however they are formed: it is the electrode reaction that is to be taken into account, and this gives the name of the particular electrode being considered. The net charge at any point is quite irrelevant to this, although it must account for external factors.
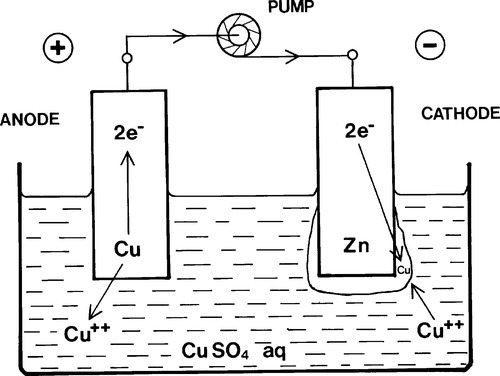
•1.5 Driving back
The same electron pump might be reversed so as to increase the net charges on the electrodes in the same sense as found in Fig. 1.4 (Fig. 1.7). At some point the electrode potential spontaneously generated would be exactly balanced and no reactions would occur, there being no net driving force. When that potential is exceeded, however, the reactions will tend to be driven in the opposite directions, Cu dissolving at the (new) anode and Cu would also be deposited at the (new) cathode (Fig. 1.7). This then is an electrolytic or plating cell. The ‘pump’, of course, is some form of battery (which strictly speaking is itself be made up of a series of spontaneous electrochemical cells) or other electrical voltage source (Fig. 1.8).
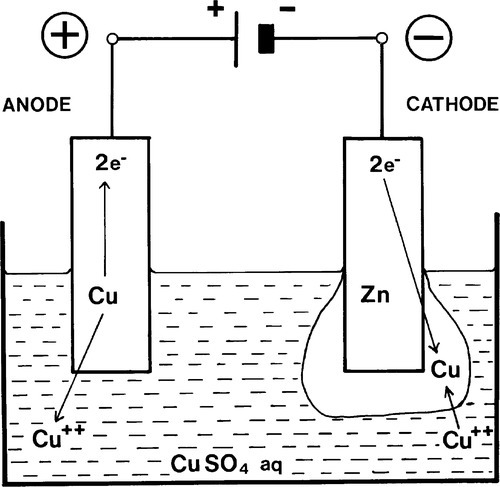
Each terminal on a dry cell, battery or other voltage source is labelled with the sign of the charge at that point, and this is therefore the same as that of the net charge residing on the attached electrode itself (Fig. 1.4). Thus the ‘positive’ terminal of a dry cell is the cathode. If you need to work out what is going on in any system, merely determine the electrode reactions, and all else follows.
§2 Polarization
Corrosion cells have been discussed as though the rate of reaction were independent of time, but this is an over-simplification in most cases. In a spontaneous cell such as that of Fig. 1.2, if the reaction is allowed to continue, the concentration of metal ions around the dissolving anode will rise. Consideration of the equilibrium that generates the potential (equation 1.3) shows that as this occurs there will be less tendency for the anode metal to dissolve, therefore a lower rate of dissolution, gradually reducing to zero. This is described as the cell becoming polarized due to the generation of a back e.m.f. (electromotive force). The back e.m.f. due to these concentration effects can eventually equal the original cell potential, resulting in an equilibrium with no further dissolution occurring. Such a closed system would be self-limiting. However, there are two major ways in which this limit may be broken, both of which are highly pertinent to dentistry.
•2.1 Limitations
The first arises because most of the metals used have rather insoluble hydroxides. This means that they react readily with water:
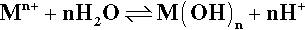
This has two effects. Primarily, the concentration of metal ions is kept down to correspond with the solubility of the hydroxide. Secondly, hydrogen ions are generated, lowering the pH, thereby increasing the solubility of the hydroxide somewhat, but also increasing the potential for dissolution. The fate of the hydrogen ions will be discussed in a moment.
The second process occurs when the system is open (as opposed to the closed systems illustrated so far), which means that as fast as metal is dissolved, the ions are carried away by diffusion or bulk flow in the electrolyte so that the ion concentration cannot build up. Corrosion in the mouth frequently occurs under such circumstances because of the flow of saliva and foodstuffs. The corrosion product ions are maintained at a low concentration in the vicinity of the corrosion site, and thus the corrosion rate remains high, very similar to the initial rate.
•2.2 Depolarization
In a closed system the build-up of metal ions in the vicinity of an anode results in an approach to equilibrium and the cessation of dissolution, limited by diffusion. In addition, the rate of deposition of metal ions at the cathode may exceed the capacity of diffusion to replace them. Depolarization may be then effected by stirring, which redistributes the solutes in the electrolyte. The potential measured across a cell such as is shown in Fig. 1.4 will be strongly dependent on such effects, and the experimental determination of cell e.m.f.s is made more difficult as the exact conditions at each electrode become difficult to control. Convection, due to changes in density resulting from the dissolving metal or temperature differences, may also cause variation in potentials over time.
§3 Cathode Processes
So far it has been assumed that certain suitable metal ions already exist in the electrolyte for the cathodic reduction reaction. This obviously will not always be the case (and especially not so in the mouth) but there may be other reactions possible to serve as electron sinks, such as:
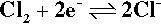
which may be relevant in swimming pools and chlorinated drinking water;
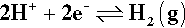
which tends to be difficult unless on specially-prepared catalytic surfaces; and
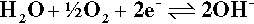
or, equivalently,
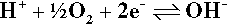
If going to the right, these are all reduction reactions, consuming electrons. The second example may be familiar as one half of the electrolysis of water, the complementary oxidation (of hydroxide to form oxygen) being the reverse of the third example.
The third example, as written, is one of the most important cathodic reactions in any dental context (and indeed in many others) because of the nearly universal presence of the very reactive gas oxygen dissolved in the electrolyte, be it saliva, blood, sterilization liquid or whatever. We can generally assume, therefore, that the cathode reaction in the oral environment is the reduction of oxygen. Note that this does not in any way affect the arguments above in terms of electrochemical cells, but merely facilitates their occurrence in a wider range of contexts. It can be seen that hydrogen ions are effectively consumed in this process and so must diffuse from the anode, so decreasing the tendency for the pH to fall there and, conversely, rise at the cathode. These pH changes provide one very simple means of detecting such electrochemical reactions and so identifying directly which electrode is which. Either by adding an appropriate pH indicator dye to the system, or by using a ‘pH electrode’ (itself a special type of electrochemical half-cell), the zones of altered pH may be visualized directly or mapped.
An alternative view of the meaning of reaction 3.3b concerns the driving force for moving to the right. It is apparent that, first, acidic conditions promote reaction. The hydro/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


