Malignant tumors of visceral organs are a fundamental feature of familial cancer and paraneoplastic syndromes. In many instances, the presence of an internal and often occult malignancy may be forewarned by various external manifestations. Several of these findings are preferentially localized to the head and neck region, including the oral cavity proper. This places the dental practitioner in a unique position to detect these “markers” of occult neoplastic involvement. Because these markers may present before an established syndrome or cancer diagnosis, even representing the first expression of disease in some cases, early recognition by a dentist may lead to timely diagnosis and management of these cancer-associated syndromes.
Malignant tumors of visceral organs are a fundamental feature of familial cancer and paraneoplastic syndromes. In many instances, the presence of an internal and often occult malignancy may be forewarned by various external manifestations. Several of these findings are preferentially localized to the head and neck region, including the oral cavity proper. This places the dental practitioner in a unique position to detect these “markers” of neoplastic involvement. Because these markers may present before an established syndrome or cancer diagnosis, early recognition on the part of the dentist may lead to timely diagnosis and management of these cancer-associated syndromes.
Orofacial markers of internal malignancy
The familial cancer syndromes are a group of inherited disorders characterized by predisposition to visceral malignancies. Many of these syndromes have oral and facial manifestations that may precede, follow, or occur synchronously with internal neoplasia. Early diagnosis of familial cancer syndromes is crucial as many of the associated malignancies are potentially curable but fatal if left untreated.
Orofacial markers of internal malignancy include (1) osteomas of Gardner’s syndrome (GS), (2) mucosal neuromas of multiple endocrine neoplasia type 2B, (3) oral and cutaneous papules of Cowden disease (CD), (4) periorificial lentigines of Peutz-Jeghers syndrome, (5) sebaceous tumors of Muir-Torre syndrome, and (6) odontogenic keratocysts of nevoid basal cell carcinoma syndrome. Other familial cancer syndromes with orofacial manifestations are listed in Table 1 .
| Syndrome | Orofacial marker | Predominant malignancies |
|---|---|---|
| Gardner’s syndrome | Osteomas; epidermoid cysts | Colorectal cancer |
| Multiple endocrine neoplasia type 2B | Mucosal neuromas | Medullary thyroid carcinoma; pheochromocytoma |
| Cowden disease | Oral and skin papules | Breast cancer; thyroid cancer |
| Peutz-Jeghers syndrome | Periorificial lentigenes | Gastrointestinal cancer |
| Muir-Torre syndrome | Sebaceous neoplasms | Colorectal cancer |
| Nevoid basal cell carcinoma syndrome | Odontogenic keratocysts; basal cell carcinomas | Central nervous system tumors (including medulloblastoma), ovarian tumors |
| Neurofibromatosis type 1 | Neurofibromas | Malignant peripheral nerve sheath tumor |
| Carney complex | Lentigines | Pituitary, testicular, and thyroid tumors |
| Birt-Hogg-Dubé syndrome | Skin fibrofolliculomas and trichodiscomas | Renal cell carcinoma |
| von Hippel-Lindau disease | Skin hemangiomas | Central nervous system tumors; renal cell carcinoma; pheochromocytoma |
Osteomas
Clinical and microscopic features
Osteomas are benign bony lesions that have a predilection for the craniofacial skeleton, particularly the mandible and condyle . Most gnathic osteomas are asymptomatic, slowly growing lesions that show early enlargement and stabilization over time . Intraorally, a mass may or may not be seen in the affected area. In contrast, involvement of the condyle often produces clinical symptoms, such as progressive deviation of the midline or limited mouth opening . Radiographically, osteomas present as well-circumscribed radiopaque masses that can be difficult to distinguish from more common osteosclerotic inflammatory conditions and benign fibroosseous lesions. Microscopic examination shows unremarkable lamellar or woven bone with variable amounts of marrow.
Gardner’s syndrome
Osteomas may occur sporadically or in the setting of GS. GS is defined by a complex of premalignant gastrointestinal polyps; osteomas; and various cutaneous, ocular, and soft tissue lesions . Osteomas are found in approximately 80% of patients who have GS , and identification of three or more jaw osteomas is considered highly suggestive of this disease ( Fig. 1 ) . They often appear before puberty and may represent the first manifestation of disease . Besides osteomas, patients who have GS may exhibit other gnathic stigmata, such as impacted and supernumerary teeth and odontomas . The cutaneous abnormalities consist predominantly of facial and scalp epidermoid cysts but may include other benign mesenchymal proliferations, such as fibromas and lipomas . The osteomas and skin lesions often precede polyposis by as many as 10 years . Hamartomas of the retinal pigment epithelium (pigmented ocular fundus lesions) are another common finding that tend to present early in disease . Together, the osteomas, epidermoid cysts, and PFOLs may serve as valuable premonitory markers of this syndrome.
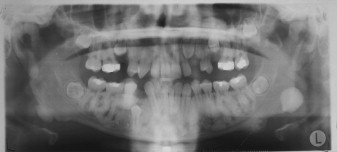
GS is a hereditary disorder that is widely regarded as a variant of familial adenomatous polyposis (FAP). All conditions within the FAP spectrum are characterized by mutations of the adenomatous polyposis coli ( APC ) gene on chromosome 5 . Most cases (75%) are inherited in an autosomal dominant fashion, whereas the remaining cases arise from de novo mutations . The APC gene itself is a tumor suppressor gene whose protein product, APC, is predominantly expressed in human epithelial cells . Studies have shown that mutational positioning within the APC gene may influence expression of disease, suggesting a specific genotypic–phenotypic correlation . Several authors have documented more severe gnathic manifestations in patients harboring mutations between codons 1444 and 1560 .
The gastrointestinal polyps in patients who have GS present early in life and often show diffuse involvement of the colon and rectum . Malignant transformation of untreated polyps to colorectal adenocarcinoma is inevitable, with a cited incidence of 100% .
Patients who have GS are also predisposed to developing locally aggressive fibrous proliferations, known as desmoid tumors . Less-common extracolonic tumors include carcinoma of the gallbladder and thyroid gland and neoplasms of the adrenal gland, liver, and central nervous system .
Management and prognosis
The osteomas themselves are completely benign. Large osteomas that are symptomatic or cosmetic concern may be treated with simple surgical excision. However, detection of multiple osteomas, especially in the setting of gastrointestinal polyps, should cause one to suspect GS. Patients should be referred for appropriate workup, including genetic analysis for FAP mutations. If the diagnosis of GS is confirmed, patients will require vigilant gastrointestinal surveillance with colonoscopy and upper gastrointestinal endoscopy . Microscopic confirmation of dysplasia or malignancy within a polyp necessitates aggressive surgical management with colon resection . Furthermore, current protocols recommend prophylactic colon resection in GS patients who have more than 30 polyps . The prognosis of colorectal adenocarcinoma in the FAP setting is poor, with metastatic disease accounting for 59% of deaths . As with all patients affected by familial cancer syndromes, genetic counseling is often warranted to identify other family members who may be at risk.
Osteomas
Clinical and microscopic features
Osteomas are benign bony lesions that have a predilection for the craniofacial skeleton, particularly the mandible and condyle . Most gnathic osteomas are asymptomatic, slowly growing lesions that show early enlargement and stabilization over time . Intraorally, a mass may or may not be seen in the affected area. In contrast, involvement of the condyle often produces clinical symptoms, such as progressive deviation of the midline or limited mouth opening . Radiographically, osteomas present as well-circumscribed radiopaque masses that can be difficult to distinguish from more common osteosclerotic inflammatory conditions and benign fibroosseous lesions. Microscopic examination shows unremarkable lamellar or woven bone with variable amounts of marrow.
Gardner’s syndrome
Osteomas may occur sporadically or in the setting of GS. GS is defined by a complex of premalignant gastrointestinal polyps; osteomas; and various cutaneous, ocular, and soft tissue lesions . Osteomas are found in approximately 80% of patients who have GS , and identification of three or more jaw osteomas is considered highly suggestive of this disease ( Fig. 1 ) . They often appear before puberty and may represent the first manifestation of disease . Besides osteomas, patients who have GS may exhibit other gnathic stigmata, such as impacted and supernumerary teeth and odontomas . The cutaneous abnormalities consist predominantly of facial and scalp epidermoid cysts but may include other benign mesenchymal proliferations, such as fibromas and lipomas . The osteomas and skin lesions often precede polyposis by as many as 10 years . Hamartomas of the retinal pigment epithelium (pigmented ocular fundus lesions) are another common finding that tend to present early in disease . Together, the osteomas, epidermoid cysts, and PFOLs may serve as valuable premonitory markers of this syndrome.
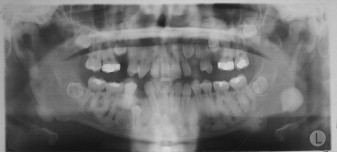
GS is a hereditary disorder that is widely regarded as a variant of familial adenomatous polyposis (FAP). All conditions within the FAP spectrum are characterized by mutations of the adenomatous polyposis coli ( APC ) gene on chromosome 5 . Most cases (75%) are inherited in an autosomal dominant fashion, whereas the remaining cases arise from de novo mutations . The APC gene itself is a tumor suppressor gene whose protein product, APC, is predominantly expressed in human epithelial cells . Studies have shown that mutational positioning within the APC gene may influence expression of disease, suggesting a specific genotypic–phenotypic correlation . Several authors have documented more severe gnathic manifestations in patients harboring mutations between codons 1444 and 1560 .
The gastrointestinal polyps in patients who have GS present early in life and often show diffuse involvement of the colon and rectum . Malignant transformation of untreated polyps to colorectal adenocarcinoma is inevitable, with a cited incidence of 100% .
Patients who have GS are also predisposed to developing locally aggressive fibrous proliferations, known as desmoid tumors . Less-common extracolonic tumors include carcinoma of the gallbladder and thyroid gland and neoplasms of the adrenal gland, liver, and central nervous system .
Management and prognosis
The osteomas themselves are completely benign. Large osteomas that are symptomatic or cosmetic concern may be treated with simple surgical excision. However, detection of multiple osteomas, especially in the setting of gastrointestinal polyps, should cause one to suspect GS. Patients should be referred for appropriate workup, including genetic analysis for FAP mutations. If the diagnosis of GS is confirmed, patients will require vigilant gastrointestinal surveillance with colonoscopy and upper gastrointestinal endoscopy . Microscopic confirmation of dysplasia or malignancy within a polyp necessitates aggressive surgical management with colon resection . Furthermore, current protocols recommend prophylactic colon resection in GS patients who have more than 30 polyps . The prognosis of colorectal adenocarcinoma in the FAP setting is poor, with metastatic disease accounting for 59% of deaths . As with all patients affected by familial cancer syndromes, genetic counseling is often warranted to identify other family members who may be at risk.
Mucosal neuromas
Clinical and microscopic features
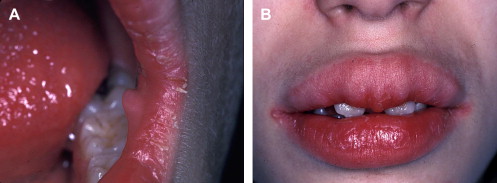
Mucosal neuromas are benign soft tissue neoplasms that present as small, painless, often pedunculated nodules. They can affect any mucosal surface but exhibit a predilection for oral mucosa, especially the tongue, lips, and commissures ( Fig. 2 A) . Microscopically, the neuromas are composed of hyperplastic nerve bundles surrounded by prominent perineural sheaths .
Multiple endocrine neoplasia
Mucosal neuromas are a hallmark of multiple endocrine neoplasia (MEN) syndrome type 2B, also known as MEN type III . MEN 2B is characterized by a clinical triad of multiple mucosal neuromas, medullary thyroid carcinoma (MTC), and pheochromocytoma . Hyperparathyroidism is a variable feature . Phenotypically, patients who have MEN 2B display a marfanoid habitus and laxity of joints . The mucosal neuromas can involve intraoral tissues and the ocular mucosa ; this gives rise to characteristic facial features, including diastema formation, everted eyelids, and protuberant lips ( Fig. 2 B). Most notably, mucosal neuromas often represent the first manifestation of this syndrome .
Approximately 95% of patients who have MEN 2B harbor a germline mutation of the rearranged during transfection ( RET ) protooncogene on chromosome 10 within the MEN 2B domain . This mutation may occur spontaneously or be inherited as an autosomal dominant trait. The RET gene encodes for the protein RET, a membrane-bound tyrosine kinase receptor involved in several cellular processes . Constitutive expression of RET is seen in cells of neural crest origin, namely the peripheral nerve cells, parafollicular cells (C cells) of the thyroid gland, and chromaffin cells of the adrenal gland . RET gene activation leads to hyperplasia and potential for neoplastic development within the target cells .
Approximately 90% of patients who have MEN 2B develop MTC, an aggressive malignancy of the thyroid parafollicular cells . Affected patients will typically show elevated calcitonin levels on blood studies.
Approximately 50% of patients will develop a neoplasm of the adrenal chromaffin cells, called pheochromocytoma , which may be bilateral or multifocal . Laboratory analysis of affected patients will show increased levels of vanillylmandelic acid and urinary catecholamines levels . Sequelae of untreated pheochromocytomas include persistent hypertension and, rarely, tumor metastasis . Other neoplasms less commonly associated with MEN 2B include gastrointestinal tumors called ganglioneuromas and tumors of the genitourinary tract .
Management and prognosis
Patients who present with multiple histologically confirmed mucosal neuromas should undergo evaluation for MEN 2B, including DNA analysis for RET mutation , biochemical testing, and targeted thyroid, adrenal, and gynecologic evaluations. MTC is associated with a universally poor prognosis because of a marked propensity for metastasis and recurrence . Because most patients who have MEN 2B develop MTC, early prophylactic therapy involving total thyroidectomy and node dissection is advocated . Any clinical enlargement of the thyroid gland during head and neck examination should prompt immediate referral to the patient’s endocrinologist for evaluation.
Oral and cutaneous papules
Clinical and microscopic features
Oral and cutaneous fibrous papules are fairly nonspecific findings that may be seen in the syndromic and nonsyndromic setting. Mucocutaneous papules can be a feature of several conditions, including but not limited to tuberous sclerosis, Heck’s disease, and CD.
Cowden disease
CD, also known as multiple hamartoma syndrome , is a rare, hereditary genodermatosis characterized by proliferations of the ectodermal, mesodermal and endodermal tissues . Patients who have CD present with numerous mucocutaneous fibrous papules and a predisposition to developing tumors of various organ systems. The mucosal or skin stigmata typically manifest by the third decade and may precede CD-associated neoplasia .
The oral papules are asymptomatic, smooth or verrucous-surfaced nodules that generally affect the gingiva, tongue, palate, and labial and buccal mucosa . Florid and confluent involvement can be seen, imparting a cobblestone appearance to the oral mucosa sometimes referred to as oral papillomatosis ( Fig. 3 A). Oral papules affect 80% to 90% of patients and represent a major element of CD . Microscopically, nonspecific fibrous and epithelial hyperplasia is seen . Additional intraoral findings that may be seen in CD include a high-arched palate and fissured tongue .
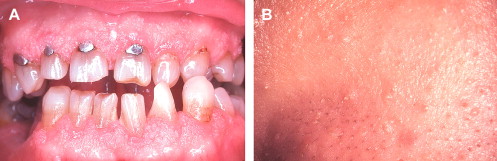
Cutaneous papules are also an integral component of the CD complex and affect an estimated 86% of patients . They typically manifest early in life and appear as dome-shaped lesions with verrucous surfaces and keratin plugging ( Fig. 3 B). A predilection for the periorificial and centrofacial skin is seen . Microscopically, the skin papules are more distinctive than their oral counterparts, with most displaying the histologic features of a benign hair follicle tumor known as a trichilemmoma . Other cutaneous findings in CD include keratosis of the hands, palms, and soles; squamous, basal, and Merkel cell carcinomas ; and a plethora of benign connective tissue tumors .
Approximately 85% of CD cases are attributable to germline mutations of the phosphatase and tensin homolog ( PTEN ) gene on chromosome 10 . This mutation may be transmitted as an autosomal dominant trait or may represent spontaneous mutation. PTEN is a ubiquitously expressed tumor suppressor gene with functions in apoptosis and cell cycle arrest; inactivation results in uncontrolled cellular proliferation . PTEN mutations are not restricted to CD but are a shared feature of the disorders collectively termed the PTEN hamartomatous tumor syndromes (PHTS).
Patients who have CD are predisposed to developing benign and malignant tumors of several organ systems . Breast disease is detected in approximately 75% of patients and consists primarily of benign fibrocystic disease and fibroadenomas. However, breast carcinoma affects 20% to 50% of female patients, and 25% of these patients are at risk for bilateral involvement . CD-associated breast malignancy often exhibits poor prognostic features, including higher grade, larger size, aggressive growth patterns, lymph node involvement, and hormone-receptor negativity . Men who have CD are also at increased risk for developing breast cancer . Benign and malignant thyroid tumors are seen in approximately 40% to 60% of patients who have CD . Genitourinary involvement has been described and can include benign ovarian cysts and fibroids, endometrial carcinoma , and, rarely, carcinomas of the cervix, bladder, and kidney . Lastly, tumors of the gastrointestinal tract and musculoskeletal and central nervous systems have also been documented.
Management and prognosis
The oral and facial papules may be removed with surgical excision, for cosmetic purposes and to rule out other cutaneous pathology. The presentation of multiple mucocutaneous papules should prompt evaluation for several clinically overlapping conditions. Timely diagnosis of CD is of paramount importance because an estimated 40% of patients will develop at least one visceral malignancy in their lifetime .
The particularly aggressive nature of CD-associated breast cancer is of greatest concern in this patient population. Therefore, all women who have CD are required to undergo lifetime mammographic follow-up with discretional biopsies. Moreover, prophylactic bilateral mastectomies have been advocated by some authorities .
Periorificial lentigines
Clinical and microscopic features
Periorificial lentigines are blue-black macules that tend to localize to the skin around the mouth, nose, eyes, anus, and genital regions . At initial presentation, the lentigines are deeply pigmented but have a tendency to fade over time . They can be distinguished from the more common ephelides (freckles), which typically vary in intensity with ultraviolet exposure and are restricted to sun-exposed areas . Microscopic examination of the lentigines shows thickened epithelium with intraepithelial dendritic melanocytes and evidence of true melanocytic hyperplasia .
Peutz-Jeghers syndrome
Periorificial lentigines are a feature of Peutz-Jeghers syndrome (PJS), a rare, autosomal dominant, familial lentiginosis also characterized by small intestine polyposis. The lentigines usually present by the first decade before the polyps develop ; congenital pigmentation has also been described . The vermillion border is affected in 95% of patients ( Fig. 4 ) , whereas intraoral extension to the labial mucosa, buccal mucosa, and tongue is seen in 66% to 83% of patients .
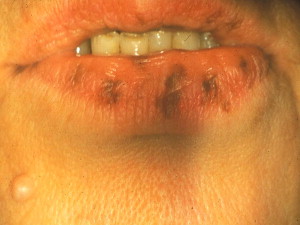
Most patients who have PJS (50%–70%) exhibit a germline mutation of the tumor suppressor gene serine/threonine kinase 11 ( STK11/LKB1 ) on chromosome 19 . Incomplete gene penetrance accounts for the marked phenotypic variability seen in PJS.
Gastrointestinal symptomology related to intestinal polyposis generally becomes apparent by the fourth decade . Although the polyps were historically considered hamartomatous, increasing evidence supports that they may harbor clonal changes and malignant potential . This finding is likely responsible for the 2% to 3% incidence of gastrointestinal adenocarcinoma in PJS . Besides gastrointestinal carcinomas, patients who have PJS are also at increased risk for developing cancers of other organ systems. Malignancies of breast, pancreas, thyroid gland, genitourinary tract, lung, and testis, among others, have been reported . Strikingly, the risk for breast cancer in PJS is comparable to women who have hereditary forms of breast cancer . PJS is also one of the strongest known risk factors for pancreatic cancer .
Management and prognosis
A diagnosis of PJS should be considered in a patient presenting with multiple periorificial and oral pigmented macules, especially in the setting of gastrointestinal symptomology or confirmed intestinal polyposis. In patients who have suspected PJS, systemic and genetic workup, including thorough gastrointestinal evaluation, is warranted. Overall, affected patients have a 10- to 18-fold increase in cancer predisposition compared with the general population . The cumulative risk for developing malignancy in PJS is age-dependent and has been reported to approach 93% by age 64 . Therefore, careful monitoring for gastrointestinal and extragastrointestinal tumors is mandatory. Ultimately, an estimated 48% of patients who have PJS will succumb to malignancy by the average age of 57 .
Sebaceous tumors
Clinical and microscopic features
Neoplasms of sebaceous origin are uncommon in the general population . These lesions appear as yellow-to-flesh–colored papules or nodules that can involve the facial skin and trunk regions . Microscopically, the spectrum of sebaceous neoplasms includes the benign entities sebaceous adenoma and sebaceous epithelioma, and the malignant tumors, basal cell carcinoma with sebaceous differentiation and sebaceous carcinoma . The discovery of multiple sebaceous tumors, especially in younger patients, should prompt further investigation for the very rare, autosomal dominant genodermatosis, Muir-Torre syndrome.
Muir-Torre syndrome
Muir-Torre syndrome is characterized by sebaceous neoplasms occurring in association with one or more visceral malignancies. Multiple keratoacanthomas may also be seen. Keratoacanthomas are microscopically distinct epithelial proliferations that have a predilection for the head and neck region and may show spontaneous regression. The cutaneous manifestations of Muir-Torre syndrome are often an early feature of this syndrome and may show synchronous or metachronous presentation with the internal malignancies. In fact, Serleth and Kisken reviewed 163 patients who had Muir-Torre syndrome and found that in 22%, skin lesions preceded the discovery of the initial internal malignancy.
Muir-Torre syndrome is currently classified as a subset of the hereditary nonpolyposis colorectal cancer (HNPCC) syndromes . Both Muir-Torre syndrome and the HNPCC conditions share a common genetic basis consisting of truncating mutations in the DNA mismatch; this predisposes to microsatellite instability and neoplastic transformation . The most common gene affected in Muir-Torre syndrome is the MSH2 gene, situated on chromosome 2 .
A large retrospective review determined that patients who have Muir-Torre syndrome develop an average of two visceral malignancies in their lifetime . The most common malignancy is colorectal carcinoma, accounting for approximately 51% of Muir-Torre syndrome–associated cancers . Malignancy of the genitourinary tract is second in frequency (25%) , followed by breast carcinoma. Isolated cases of leukemia and lymphoma, head and neck carcinomas, and lung malignancies have also been documented .
Management and prognosis
The sebaceous tumors themselves should be removed for microscopic confirmation. The unusual finding of multiple sebaceous tumors, with or without keratoacanthomas, is highly suggestive of Muir-Torre syndrome. Because patients who have Muir-Torre syndrome continue to be at risk for developing subsequent malignancies throughout their lifetime, management must include surveillance for systemic neoplasia in addition to surgical or pharmacologic treatment of the skin lesions. Most malignancies associated with Muir-Torre syndrome are low-grade and clinically indolent . Therefore, patients tend to have favorable prognoses with long-term survival .
Odontogenic keratocysts
Clinical and microscopic features
Odontogenic keratocysts (OKCs) are cystic lesions of odontogenic epithelial origin that occur exclusively within the gnathic bones, with rare examples showing extra-osseous localization. Most lesions are asymptomatic, although pain and swelling can be observed in larger cases . Radiographically, OKCs present as unilocular or more classically, multilocular radiolucent lesions that exhibit smooth and corticated borders . They may or may not be associated with teeth and can arise at any location within the jawbones, although a predilection for the mandible is seen . Microscopically, OKCs are lined with a thin layer of stratified squamous epithelium characterized by palisaded basal cells and a corrugated, parakeratotic luminal surface .
Nevoid basal cell carcinoma syndrome
The presence of multiple OKCs in a patient should raise the suspicion for nevoid basal cell carcinoma syndrome (NBCCS), also known as Gorlin’s syndrome. Patients who have NBCCS are affected by multiple OKCs, basal cell carcinomas, developmental abnormalities, and neoplasms of the neurologic, skeletal, and extraskeletal systems . Besides basal cell carcinomas, patients who have NBCCS may also have other cutaneous manifestations, such as milia, epidermal cysts, and palmar/plantar pitting . Common developmental skeletal findings include calcification of the falx cerebri and rib and vertebral anomalies . Characteristic facial features, such as macrocephaly, prominent frontal bossing, and hypertelorism , can also be seen.
The almost universal features of OKCs and basal cell carcinomas appear early in life, typically by the first and second decades, respectively . OKCs may represent the first manifestation of disease . The incidence of OKCs in the NBCCS setting has been estimated to be 74% , and patients develop an average of five lesions in their lifetime . Radiographically, syndrome-associated lesions do not differ significantly from their sporadic counterparts, with the exception of multifocal involvement in the former ( Fig. 5 A). Histologically, additional features, such as epithelial budding and daughter cysts, may be seen with increased frequency . The basal cell carcinomas associated with NBCCS are also similar, clinically and histologically, to sporadically occurring lesions . They appear as flesh-colored papules, with or without surface ulceration, that tend to localize to the midfacial and periorbital regions ( Fig. 5 B) . Involvement is typically not restricted to sun-exposed skin in syndromic cases.
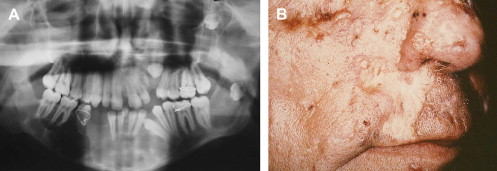
NBCCS is an autosomal dominantly inherited condition caused by germline mutation of the tumor suppressor gene patched ( PTCH ) on chromosome 9 . The developmental defects and tumor predisposition in this syndrome seem to be related to haploinsufficiency (inheritance of a defective gene) and losses of heterozygosity within the PTCH gene. Recent cytogenetic studies have identified these PTCH mutations within OKCs and basal cell carcinomas .
Neoplasms of several organ systems are a documented feature of NBCCS. Benign ovarian tumors known as ovarian fibromas occur in approximately 25% of female patients and are typically bilateral, calcified, and asymptomatic ; ovarian malignancies, including fibrosarcomas and leiomyosarcomas, have also been described . A malignant cerebellar tumor known as medulloblastoma can be seen in 5% of patients . Other tumors of cerebral origin, such as meningiomas and astrocytomas, have also been documented . Malignancies of the musculoskeletal system, including rhabdomyosarcomas and soft tissue leiomyosarcomas , have been reported. Patients who have NBCCS are also at risk for developing cardiac fibromas , which may result in hemodynamic or conduction abnormalities .
Management and prognosis
The presentation of multiple OKCs in conjunction with multiple basal cell carcinomas is highly suggestive of NBCCS, and genetic evaluation is warranted. Treatment of the OKCs consist of thorough enucleation followed by peripheral ostectomy or chemical cauterization of the bony crypt; decompression and subsequent enucleation can also be performed for larger lesions . Evidence also suggests that the recurrence potential for syndrome-associated OKCs may be higher than in sporadic lesions ; moreover, patients are at risk for developing additional OKCs throughout their lifetime. Curettage is the preferred treatment modality for the basal cell carcinomas, although recurrent lesions may be more amendable to Moh’s micrographic surgery. As with OKCs, patients will typically develop skin lesions throughout life. Unfortunately, repeated treatment for multiple OKCs and basal cell carcinomas may ultimately result in some facial disfigurement.
Patients who have confirmed NBCCS should be placed under close dental and dermatologic surveillance. Periodic neurologic assessment is recommended in patients until 7 years of age to evaluate for medulloblastomas; cardiac and gynecologic evaluations should be also undertaken if clinically indicated . The prognosis of patients who have NBCCS is favorable, although deaths have been attributed to invasion of vital structures by aggressive basal cell carcinomas, and to other internal malignancies . Development of secondary neoplasms is particularly concerning, especially in cases where medulloblastomas and basal cell carcinomas have been irradiated . Moreover, radiation of NBCCS-related medulloblastomas has resulted in early onset and increased numbers of basal cell carcinomas within the treatment field .
Paraneoplastic syndromes of the oral and facial region
Paraneoplastic syndromes (PNSs) are a group of heterogeneous disorders that represent the clinical manifestations of indirect and remote effects produced by tumor metabolites . Often the course of the PNS parallels the course of the internal malignancy; therefore, following the syndrome itself may help monitor tumor evolution, progression, or recurrence. Because the clinical presentation of a PNS can often be overwhelming, clinicians may not recognize the need to explore an underlying cause, which may have fatal consequences in the case of a primary malignancy . In other instances, the effects of the PNS itself may be more serious than the tumor and can also lead to the demise of the patient.
The most common PNSs with manifestations in the oral, head, and neck regions include paraneoplastic pemphigus, acanthosis nigricans, multiple eruptive seborrheic keratoses, herpes zoster, and Sweet syndrome. Other PNSs are listed in Table 2 .


