Composite dental restorative materials have advanced considerably over the past 10 years. Although composites have not totally replaced amalgam, they have become a viable substitute in many situations. Problems still exist with polymerization contraction stress, large differences in the coefficient of thermal expansion (CTE) of composites compared with tooth structure, and with some technique sensitivity; however, new expanding resins, nanofiller technology, and improved bonding systems have the potential to reduce these problems. With increased patient demands for esthetic restorations, the use of direct filling composite materials will continue to grow. The one major caveat to this prediction is that clinicians must continue to use sound judgment on when, where, and how to use composite restoratives in their practices.
A generalized definition of a composite is a multiphase material that exhibits the properties of both phases where the phases are complimentary, resulting in a material with enhanced properties . The materials discussed in this article are composites by definition that are used as direct esthetic restorative materials.
The first tooth-colored composite was silicate cement, which was introduced in 1870s. This composite formulation was based on alumino-fluro-silicate glasses and phosphoric acid. The dispersed phase was residual glass particles, and the matrix phase was the aluminum phosphate salt formed from the partial acid dissolution of the glass particles; however, these were brittle, required mechanical retention, and had an average longevity of only a few years .
The first polymeric tooth-colored composite used in dentistry was based on poly(methylmethacrylate). This material was developed in the 1940s, and consisted of a poly(methylmethacrylate) powder, methyl methacrylate monomer, benzoyl peroxide, and n,n-dimethlyparatoluidine. These materials could be classified as composites, because upon mixing, the polymer powder formed a dispersed phase and the monomer polymerized to form the continuous phase. The polymerization was initiated at room temperature, using the redox initiator combination of benzoyl peroxide and n,n-dimethlyparatoluidine. Although these materials were initially esthetic, they were plagued with a variety of problems, including poor color stability, high polymerization shrinkage, a lack of bonding to tooth structure, and a large coefficient of thermal expansion (CTE) .
The first polymer matrix composite incorporating silica fillers was introduced in the 1950s. These composites had improved mechanical properties and good esthetics; they did not bond to tooth structure, and still exhibited significant polymerization shrinkage. In addition, there was no significant bonding between the silica particles and the polymer matrix. Consequently, these composites did not have good wear resistance clinically, because the filler particles were easily dislodged . New improved formulations incorporated a coupling agent such as γ-methacryloxpropyl-trimethoxy silane or vinyl triethoxysilane. The coupling agent provided a method to covalently bond the filler particles to the resin matrix. The resulting composite exhibited improved mechanical properties and wear resistance; however, the polymerization shrinkage and lack of bonding to tooth structure limited the clinical success of these formulations.
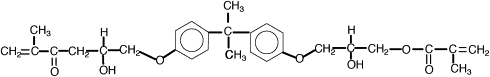
One way to address the polymerization shrinkage problem is to use high molecular weight monomers. In 1962 Bowen , while at the National Bureau of Standards, synthesized an acrylated epoxy using glycidylmethacrylate and Bisphenol A epoxy for use as a matrix for dental composite. The resulting monomer, called Bis-GMA or Bowen’s resin, possessed the viscosity of honey, and therefore limited the amount of filler particles that could be incorporated. Subsequent experiments incorporated triethylene glycol dimethacrylate (TEGDMA) as a diluent to reduce the viscosity. This monomer combination worked well, and has become one of the most widely used matrix monomer combinations for dental composites to date. The structures of Bis-GMA and TEGDMA are shown in Figs. 1 and 2 , respectively. Both of these monomers contain two reactive double bonds, and when polymerized, form covalent bonds between the polymer chains known as cross-links. Cross-linking improved the properties of the matrix phase, and the composite produced had improved mechanical and physical properties . Additional composite formulations have been prepared using various diluent monomers such as methyl methacrylate (MMA) and ethylene glycol dimethacrylate (EGDMA), and an additional high molecular weight monomer based on a urethane dimethacrylate (UDMA). The chemical structure for UDMA is illustrated in Fig. 3 .
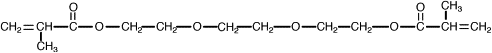
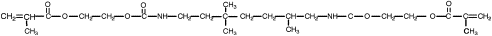
Additional monomers based on poly-acid modified acrylates have been used to formulate composites called compomers. The fillers used in compomers are silicate-based glasses and sodium fluoride. They are polymerized using free radical chemistry initiated by photoactive species or redox initiator systems. These materials were designed to have the handling properties of a traditional resin composite and the fluoride-releasing properties of a glass ionomer. Because of the hydrophilic nature of the resins, compomers actually absorb fluid from the oral environment, causing an expansion of the composite that offsets a portion of the polymerization shrinkage which occurs during setting. Compomers do not have the mechanical properties of more traditional composites, or the amount of fluoride release of glass ionomers, but have been used successfully as a direct restorative resin in some applications .
Composite fillers
The reinforcing phase in direct dental restoratives is based on glass or ceramic particles. Incorporation of these inorganic particles imparts improved strength and wear properties, decreased CTE, and reduced polymerization shrinkage. In addition, incorporation of heavy metals into the filler provide radiopacity. The initial composite fillers were limited in size because of the limited ability to grind and sieve quartz, glass, borosilicate, or ceramic particles. The particle size range was from 0.1 to 100 μm. Smaller particles have been prepared through hydrolysis or precipitation to produce what is termed fumed or pyrolitic silica. The particle sizes obtained from this process range from 0.06 to 0.1 μm .
The most recent process to form particles is through sol-gel chemistry, which uses silicate precursors that are polymerized to form particles ranging from nm to μm dimensions . This sol-gel process can be used to form almost mono dispersed particle sizes, which can be a significant advantage because different particle sizes can be produced and blended to optimize the packing efficiency and filler loading of the composite. In addition, the ability to produce submicron size particles allows the production of nanocomposites in which the particles approach the size of the polymer matrix molecules. Theoretically, nanocomposites have the potential to exhibit excellent mechanical and physical properties at higher filler loadings .
Composite resin chemistry
To reduce polymerization shrinkage and increase mechanical and physical properties requires the use of high molecular weight monomers that have the ability to cross-link. The high molecular weight reduces the volume change during polymerization. Cross-linking forms covalent bonds between the polymer chains, resulting in a dramatic increase in modulus and reduction in solubility . Bowen’s resin is the reaction product between Bisphenol A and glycidyl dimethacrylate. The chemical name is 2,2-bis[4-(2 hydroxy-3 methacryloxy proproxy)-phenyl]-propane, but it is commonly referred to as Bis-GMA. This long-chain monomer is multifunctional, having two methacrylate groups that allow it to cross-link during polymerization; however, because of its large size, Bis-GMA is highly viscous, and limits the ability to formulate composites having high filler loadings. Consequently, a lower molecular weight monomer such triethylene glycol dimethacrylate (TEGDMA) or EDMA is added to reduce the viscosity and allow increased filler loadings to be used. These monomers are also multifunctional and increase the number of cross-linking reactions during setting of resin matrix. These lower viscosity monomers may comprise 10% to 50% of a composite’s composition.
Although these monomers allow increased filler concentrations, their incorporation can lead to greater polymerization shrinkage. In addition, these monomers can produce composites with increased flexibility and decreased abrasion resistance. It has been suggested that these low molecular weight monomers increase the time before gelation of the matrix occurs, and subsequently reduce marginal polymerization contraction stress . Consequently, incorporation of these monomers can have both positive and negative effects on the composite’s properties.
One of the most significant problems with current monomers used for direct composite restorative materials is the shrinkage that occurs during polymerization. Currently, all commercial dental composites are based on vinyl monomers polymerized using free radical initiators. Conversion of these monomers results in a decrease in distance between the molecules, from a Van der Waals gap to the distance of a covalent bond. Although this distance is very small for a single monomer, the distance change over a long polymer chain is significant. Inclusion of filler reduces the volume of resin and its volume change, but the amount of filler incorporation is approaching the maximum theoretical packing fraction of 74 volume % for close-packed structures . The amount of shrinkage is controlled by the volume of resin, its composition, and the degree of conversion. Current commercial dental composites have a volumetric shrinkage ranging from 1.6 to 8 volume % . The contraction stress developed at the margin of the restoration can be sufficient to overcome the bond strength of the bonding system, resulting in a contraction gap . The contraction gap can lead to microleakage and all its associated problems (eg, secondary caries and pain).
In a recent study, the contraction stress was measured to range from 3.3 to 23.5 Mpa . During polymerization at room temperature, the resin matrix gels, and the polymer formed is below its glass transition temperature (Tg). Therefore, the amount of flow available to the polymer matrix to relieve the contraction stress during polymerization is limited. Low molecular weight diluent resins can provide more flow, but have the potential to reduce the mechanical properties of the matrix. The relationship between contraction stress and composite composition is complex, but Kleverlann and Feilzer did find correlations between volumetric shrinkage and contraction stress that suggest that lower amounts of shrinkage in current highly filled composites actually result in higher contraction stress. This surprising result may be related to the ability of formulations that contain higher concentrations of low molecular weight monomers being able to reduce contraction stress by molecular relaxations and flow.
One approach to reduce polymerization shrinkage and contraction stress is through the development of low-shrinkage or expanding monomer systems. These resin systems are based on ring-opening polymerization reactions that do not shrink to the extent of conventional vinyl polymerization resins. Monomers based on spiro-ortho carbonate have been prepared and evaluated in composite formulations. Although the composites formulated using these monomers did show less polymerization shrinkage, the property improvements were only incremental, and probably not significant enough to be realized clinically .
Eick and colleagues have reported composite matrix resins based on cycloaliphatic expoxies. These resins are polymerized using photoactive cationic initiators. Although the resins show promise, they require further optimization before a commercial product can be produced.
Chen and colleagues have developed another expoxy-based resin matrix, and prepared a composite that also incorporated nanofiller technology. The composite exhibited mechanical and physical properties that are comparable to commercial dental composites, and the strain measured during polymerization was significantly reduced. Based on these studies, commercialization of low-shrinkage monomers may be realized in the near future. The development of low-shrinkage or expanding monomers could be one of the most significant advances in direct dental restorative materials since their introduction.
One problem that has not been addressed is the large difference between the CTE of resin composites and tooth structure. The CTE of tooth structure ranges from 9 to 11 ppm/°C, compared with 28 to 50 ppm/°C for dental composite restoratives . The differential expansion and contraction of composites cause additional stress at the margin of the restoration that contribute to fatigue failure of the bond between the composite and tooth structure. Currently the only way to lower the CTE of composites is to increase the filler loading.
Composite resin chemistry
To reduce polymerization shrinkage and increase mechanical and physical properties requires the use of high molecular weight monomers that have the ability to cross-link. The high molecular weight reduces the volume change during polymerization. Cross-linking forms covalent bonds between the polymer chains, resulting in a dramatic increase in modulus and reduction in solubility . Bowen’s resin is the reaction product between Bisphenol A and glycidyl dimethacrylate. The chemical name is 2,2-bis[4-(2 hydroxy-3 methacryloxy proproxy)-phenyl]-propane, but it is commonly referred to as Bis-GMA. This long-chain monomer is multifunctional, having two methacrylate groups that allow it to cross-link during polymerization; however, because of its large size, Bis-GMA is highly viscous, and limits the ability to formulate composites having high filler loadings. Consequently, a lower molecular weight monomer such triethylene glycol dimethacrylate (TEGDMA) or EDMA is added to reduce the viscosity and allow increased filler loadings to be used. These monomers are also multifunctional and increase the number of cross-linking reactions during setting of resin matrix. These lower viscosity monomers may comprise 10% to 50% of a composite’s composition.
Although these monomers allow increased filler concentrations, their incorporation can lead to greater polymerization shrinkage. In addition, these monomers can produce composites with increased flexibility and decreased abrasion resistance. It has been suggested that these low molecular weight monomers increase the time before gelation of the matrix occurs, and subsequently reduce marginal polymerization contraction stress . Consequently, incorporation of these monomers can have both positive and negative effects on the composite’s properties.
One of the most significant problems with current monomers used for direct composite restorative materials is the shrinkage that occurs during polymerization. Currently, all commercial dental composites are based on vinyl monomers polymerized using free radical initiators. Conversion of these monomers results in a decrease in distance between the molecules, from a Van der Waals gap to the distance of a covalent bond. Although this distance is very small for a single monomer, the distance change over a long polymer chain is significant. Inclusion of filler reduces the volume of resin and its volume change, but the amount of filler incorporation is approaching the maximum theoretical packing fraction of 74 volume % for close-packed structures . The amount of shrinkage is controlled by the volume of resin, its composition, and the degree of conversion. Current commercial dental composites have a volumetric shrinkage ranging from 1.6 to 8 volume % . The contraction stress developed at the margin of the restoration can be sufficient to overcome the bond strength of the bonding system, resulting in a contraction gap . The contraction gap can lead to microleakage and all its associated problems (eg, secondary caries and pain).
In a recent study, the contraction stress was measured to range from 3.3 to 23.5 Mpa . During polymerization at room temperature, the resin matrix gels, and the polymer formed is below its glass transition temperature (Tg). Therefore, the amount of flow available to the polymer matrix to relieve the contraction stress during polymerization is limited. Low molecular weight diluent resins can provide more flow, but have the potential to reduce the mechanical properties of the matrix. The relationship between contraction stress and composite composition is complex, but Kleverlann and Feilzer did find correlations between volumetric shrinkage and contraction stress that suggest that lower amounts of shrinkage in current highly filled composites actually result in higher contraction stress. This surprising result may be related to the ability of formulations that contain higher concentrations of low molecular weight monomers being able to reduce contraction stress by molecular relaxations and flow.
One approach to reduce polymerization shrinkage and contraction stress is through the development of low-shrinkage or expanding monomer systems. These resin systems are based on ring-opening polymerization reactions that do not shrink to the extent of conventional vinyl polymerization resins. Monomers based on spiro-ortho carbonate have been prepared and evaluated in composite formulations. Although the composites formulated using these monomers did show less polymerization shrinkage, the property improvements were only incremental, and probably not significant enough to be realized clinically .
Eick and colleagues have reported composite matrix resins based on cycloaliphatic expoxies. These resins are polymerized using photoactive cationic initiators. Although the resins show promise, they require further optimization before a commercial product can be produced.
Chen and colleagues have developed another expoxy-based resin matrix, and prepared a composite that also incorporated nanofiller technology. The composite exhibited mechanical and physical properties that are comparable to commercial dental composites, and the strain measured during polymerization was significantly reduced. Based on these studies, commercialization of low-shrinkage monomers may be realized in the near future. The development of low-shrinkage or expanding monomers could be one of the most significant advances in direct dental restorative materials since their introduction.
One problem that has not been addressed is the large difference between the CTE of resin composites and tooth structure. The CTE of tooth structure ranges from 9 to 11 ppm/°C, compared with 28 to 50 ppm/°C for dental composite restoratives . The differential expansion and contraction of composites cause additional stress at the margin of the restoration that contribute to fatigue failure of the bond between the composite and tooth structure. Currently the only way to lower the CTE of composites is to increase the filler loading.
Curing of dental composites
The majority of current dental composites are cured using visible light ranging from 450 to 475 nm. Light sources include quartz halogen, laser, plasma arc, and most recently, light emitting diodes (LED). The minimum energy required for adequate curing is 300 mW/cm 2 . Newer lights have incorporated curing modes that step or ramp up the light intensity with time. These modes were added in an attempt to control the polymerization shrinkage and reduce the polymerization contraction stress. Although these lights have shown some promise, the clinical effectiveness of these controlled polymerization techniques is unknown. All of the lights used for curing composite increase the temperature of the composite to varying extents, which can actually increase the degree of conversion ; however, high-intensity light sources may cause sufficient temperature increases to result in damage to the pulp.
Composite classification: properties and applications
There has been a number of classification systems proposed to describe composite restoratives. One of the most often used classification systems is based upon filler particle size. As composite restoratives have evolved, the size of filler particles and their size distribution have been changed, in an attempt to achieve the best possible mechanical properties while maintaining esthetics. This discussion uses the following broad classifications: microfills, hybrids, packables, and compomers. In addition, subclassifications, including flowables, and nano- and microhybrids, are addressed. A summary of some of the characteristics for direct composite restoratives is given in Table 1 .
| Filler content | ||||
|---|---|---|---|---|
| Composite classification | Weight % | Volume % | Volume shrinkage (%) | Average particle size (μm) |
| Hybrid | 74–87 | 57–72 | 1.6–4.7 | 0.2–3.0 |
| Nanohybrid | 72–87 | 58–71 | 2.0–3.4 | 0.4–0.9 (macro) |
| — | — | — | 0.015–0.05 (nano) | |
| Microfills | 35–80 | 20–59 | 2–3 | 0.04–0.75 |
| Flowables | 40–60 | 30–55 | 4–8 | 0.6–1.0 |
| Compomers | 59–77 | 43–61 | 2.6–3.4 | 0.7–0.8 |
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


