8
Hematological Disease
I. Background
This chapter addresses diseases of the white blood cells (leukemias, lymphomas, multiple myeloma [MM]), red blood cells (RBCs) (anemias), and patients requiring hematopoietic stem cell (bone marrow) transplants (HSCTs).
Description of Disease/Condition
Leukemia
Leukemia refers to a group of hematological malignancies characterized by the abnormal proliferation or increased life span of immature white blood cells in the bone marrow and peripheral blood.
Leukemia can be divided into two main types: lymphocytic leukemia and myeloid leukemia. Lymphoid precursor cells developing in lymphatic tissue give rise to T lymphocytes and B lymphocytes. Myeloid cells are derived from the bone marrow and differentiate into erythrocytes, polymorphonuclear leukocytes, monocytes, eosinophils, basophils, and platelets.1 Of these main types, there are various subtypes of leukemia based on the predominant malignant cell/precursor cell type seen. Leukemia is also classified depending on duration and onset of diseases as acute or chronic.2
Acute Lymphocytic Leukemia (ALL)
- It is often an early childhood disease with the mean age of occurrence being 2–4 years.
- It is the most common leukemia in children, but can be seen at any age.
- Most cases involve B-cell malignant proliferation, followed by T-cell lymphocytic leukemia.
- Proliferating immature lymphoblasts may accumulate in the lymph nodes, liver, and spleen, in addition to peripheral blood and bone marrow.3
Chronic Lymphocytic Leukemia (CLL)
- It is characterized by the abnormal proliferation of certain types of lymphocytes, mostly CD5+ B lymphocytes.2 CD refers to a specific marker known as cluster of differentiation. CD 5 is found on cell surfaces of T and B lymphocytes.
- It can manifest with little or no signs or symptoms other than mild lymphadenopathy.
- It may often be an incidental finding during a routine complete blood count (CBC).
Acute Myeloid Leukemia (AML)
- It involves uncontrolled proliferation of clonal myelocytic leukocytes in the bone marrow and peripheral circulation.
- An accepted guideline for reaching a diagnosis of AML is the presence of at least 30% blast cells in peripheral blood.2
Chronic Myeloid Leukemia (CML)
- It is the neoplasm of mature myeloid leukocytes.
- Genetic basis is the reciprocal translocation (referred to as the Philadelphia chromosome) of the cellular gene ABL from chromosome 9 to the BCR gene on chromosome 22.2
Anemia
Anemia is defined as the decrease in the oxygen-carrying capacity of blood. Anemia can be caused by either a decreased production of RBCs, increased destruction of RBCs, increased demand for iron, or formation of abnormal erythrocytes in place of physiological cells. Normal hemoglobin consists of two pairs of globin chains (α and β, δ or γ).
Based on the underlying etiopathogenesis, anemias are classified as:
- blood loss anemia;
- iron-deficiency anemia;
- anemia of chronic disease4;
- hemolytic anemias: glucose-6 phosphate dehydrogenase deficiency (G6PD)-induced nonimmune or autoimmune anemias;
- hemoglobinopathies such as sickle cell anemia (SCA) or thalassemia5;
- hypoproliferative anemias: folate- or B12-deficiency anemia, pernicious anemia, aplastic anemia (AA).
Anemias can also be classified based on size of RBCs as:
- microcytic (mean corpuscular volume <80 micron): iron deficiency, thalassemia;
- normocytic (mean corpuscular volume 80–100 micron): SCA, G6PD deficiency, AA, blood loss anemia;
- macrocytic (mean corpuscular volume >100 micron): pernicious anemia, folate deficiency, B12 deficiency.
Iron-Deficiency Anemia
- This is the most common type of anemia.
- It is classified as a microcytic anemia.
- It is caused by low iron ingestion, excessive blood loss, or increased demand for iron.
G6PD Anemia
- It is associated with an inherited deficit of the enzyme G6PD that is needed for the biochemical hexose monophosphate (HMP) shunt pathway. On erythrocytes, it facilitates the conversion of carbohydrates into energy.
- It is classified as a normocytic hemolytic-type anemia associated with the increased rate of destruction of RBCs in response to oxidative stress.
SCA
- It is classified as an inherited hemoglobinopathy resulting from structurally deficient hemoglobin protein, which impedes its oxygenation capacity and ability to circulate throughout capillary beds. Typical hemoglobin levels are 6–9 g/dL in SCA.5
Thalassemia
- It is classified as a hemoglobinopathy sometimes manifesting as a microcytic hemolytic anemia.
- Thalassemias are a group of hereditary disorders resulting from decreased production of globin chains of the hemoglobin molecule.
Vitamin B12– and Folate-Deficiency Anemia
- It is classified as hypoproliferative macrocytic anemia.
- Together they constitute megaloblastic anemia.
- Megaloblastic anemia refers to the anemic state wherein RBCs are macrocytic, or larger than normal.
AA
- This is a rare condition resulting in complete suppression of bone marrow and the consequent depletion of all hematopoetic cell lines.6
Lymphoma
Lymphoma refers to a group of heterogeneous malignancies of lymphoid tissue or precursors of lymphoid tissue in the body. Lymphomas occur across the spectrum of lymphoid cell types—B cell, T cell, mucosa-associated lymphoid tissues (MALTs) and natural killer (NK) cells. The most common classifications of lymphomas are Hodgkin’s lymphoma and non-Hodgkin’s lymphoma (NHL).
Hodgkin’s Lymphoma
- This is a lymph node neoplasm involving overproliferation of B lymphocytes.
- The defining histological feature is the presence of a distinct cell type called Reed–Sternberg giant cell.2
NHL
- Most cases of NHL involve B cells, followed by T-cell neoplasms and NK cell neoplasms.
- While NHL is a widespread disease and can involve multiple organ systems, initial stages of the malignancy have a favorable prognosis.2
Multiple Myeloma (MM)
- This is a malignancy involving the overproliferation of abnormal immunoglobulin-secreting plasma cells.
- The consequent abnormal immunoglobulin fragments accumulate in the bone marrow.
- Sequelae of MM can be osteolysis, bone marrow suppression, bleeding disorders, and renal dysfunction.7
Pathogenesis/Etiology
Leukemia
- ALL: Pathogenesis involves excessive proliferation and circulation of immature lymphoblasts as opposed to the normal ratio of blood elements. Causes remain speculative. Environmental, genetic, viral and infectious etiologies have been entertained as possible causes. ALL is more frequent in patients with Down syndrome, and increased detection of the Philadelphia chromosome has been linked with ALL.8
- CLL: As in ALL, there is an overproliferation of immature lymphocytes, but at a slower rate. Although unknown, inheritance and exposure to environmental carcinogens have been postulated as potential causes of CLL.9
- AML: AML exhibits a rapid onset of increased numbers of immature myeloid blast leukocytes in circulation. Underlying genetic or environmental mechanisms leading to an uncontrolled proliferation of myeloid cells remain unclear, but AML occurrence in younger people is often spontaneous. In the elderly, it may arise as a sequel to a myelodysplastic state of the bone marrow.2,3
- CML: Blast phase comprises over 20–30% leukemic cells in the bone marrow. The exact mechanism triggering the genetic translocation that leads to CML is unknown but some association has been claimed to radiation exposure and chemicals.
Anemia
- Iron-Deficiency Anemia: Decreased availability of iron impedes normal erythrocytosis, and resultant systemic effects of the disease are a consequence of tissue hypoxia:
- Low iron consumption is associated with poor socioeconomic strata and poor dietary habits.
- Excessive blood loss due to
- heavy menses in women;
- slow blood loss as seen in malignant gastrointestinal ulcers.
- Increased iron demand due to
- parturition or pregnancy in women;
- sequel of chronic diseases:
- autoimmune diseases (systemic lupus erythematosus [SLE], Crohn’s disease, rheumatoid arthritis, or ulcerative colitis);
- chronic liver disease (hepatitis or cirrhosis);
- hematological malignancies (leukemias and lymphomas).10
- G6PD Anemia: Affected individuals have a faulty HMP shunt during the glycolytic pathway of glucose metabolism in RBCs thereby impeding RBCs’ ability to manage an oxidative state.10 The obstructed HMP shunt pathway leads to the accumulation of toxic oxidants inside RBCs that result in methemoglobinemia and aggregate to form Heinz bodies. Heinz bodies circulate through the spleen and liver with difficulty and are removed through hemolysis. Hemolytic episodes in affected individuals can be triggered by the exposure of the bloodstream to oxidative substances such as drugs (sulfonamides, antimalarials, aspirin, dapsone, phenacetin, and vitamin K), fava beans, and during infectious states of the body.
- SCA: At decreased pH or lowered oxygen tension, RBCs with this defective structure become sickle shaped. Sickle-shaped RBCs are rigid, eventually leading to stasis of blood flow. See Fig. 8.1. Sluggishly flowing blood accumulates in the spleen and erythrocytes are destroyed by hemolysis. This slow-circulating blood further depletes oxygen tension, leading to further sickling of RBCs—this vicious cycle can cause sickle-cell crisis in patients. Sickle cell crisis can be the consequence of oxygen-lowering states of the body such as dehydration, hypoxia, acidosis, or hypotension. SCA is an autosomal recessive disorder that needs the presence of both copies of the recessive gene (homozygous state) to manifest itself. Sickle-cell trait on the other hand does not result in the disease and is a heterozygous state involving one defective gene and a functional gene together in a pair. See Fig. 8.2. The structural deficiency of SCA stems from the substitution of a valine group with glutamic acid on position 6 of the beta chain of hemoglobin.10
- Thalassemia: In thalassemia major, at the cellular level, RBCs become more permeable secondary to aggregation of excessive number of defective globin chains. These RBCs are removed from circulation by phagocytosis or hemolysis. Compensatory production of RBCs occurs by expansion of bone marrow compartments and extramedullary erythropoiesis, but is usually insufficient. In alpha-thalassemia there is a depleted production of alpha chains. Beta-thalassemia is characterized by either a decreased production of beta-globin chains or a complete absence of these chains based on the gene mutations involved. Beta-thalassemia minor patients are heterozygous individuals who carry the trait while homozygous individuals have beta-thalassemia major or Cooley’s anemia, a fulminant type of congenital hemolytic anemia. Across the spectrum from thalassemia minor to major symptoms may vary from presence of mild or no symptoms in thalassemia minor to profound hemolytic anemia and its accompanying manifestations.10
- Vitamin B12– and Folate-Deficiency Anemia: Vitamin B12 and folic acid are both vital ingredients for erythrocytosis within the bone marrow. A deficiency in either of these components results in decreased production of erythrocytes, resulting in anemia. Strict B12 deficiency is rare and occurs in those observing an exclusive vegetable-based diet. B12 deficiency occurs most commonly secondary to deficiency of intrinsic factor in the gastrointestinal tract, and is referred to as pernicious anemia. Intrinsic factor in the gastric mucosa binds to vitamin B12, protecting the latter from proteolysis and facilitating its migration across the ileal mucosa for absorption. Vitamin B12 deficiency may also occur as a side effect of certain medications, celiac disease, sprue, or Crohn’s disease. Folic acid deficiency is observed in individuals whose diets are poor in leafy vegetables and fruits or with faulty absorption of folic acid, such as in chronic alcoholics and substance abusers. Rarely, genetic defects in folate metabolism may cause folate-deficiency anemia. Absorption of folate is also impaired as a result of cancer chemotherapy drugs.10
- AA: Bone marrow suppression as seen in AA causes significant leukopenia and anemia and their sequelae.6 This severe pancytopenia observed in otherwise healthy adults has a rapid onset and progression. AA is an idiopathic condition. Chemicals, radiation, chemotherapy, and autoimmune diseases have been implicated in some cases as etiological agents of AA. While true AA is a disease of young adulthood and old age, a rare autosomal recessive disease called Fanconi’s anemia has an earlier age of onset affecting infants and young children.11
Figure 8.1 (A) The figure shows normal red blood cells flowing freely in a blood vessel. The inset image shows a cross section of a normal red blood cell with normal hemoglobin. (B) This shows abnormal, sickled red blood cells blocking blood flow in a blood vessel. The inset image shows a cross section of a sickle cell with abnormal (sickle) hemoglobin forming abnormal strands.
Source: National Heart, Lung, and Blood Institute; National Institutes of Health; U.S. Department of Health and Human Services. Available at: http://www.nhlbi.nih.gov/health/health-topics/topics/sca/.
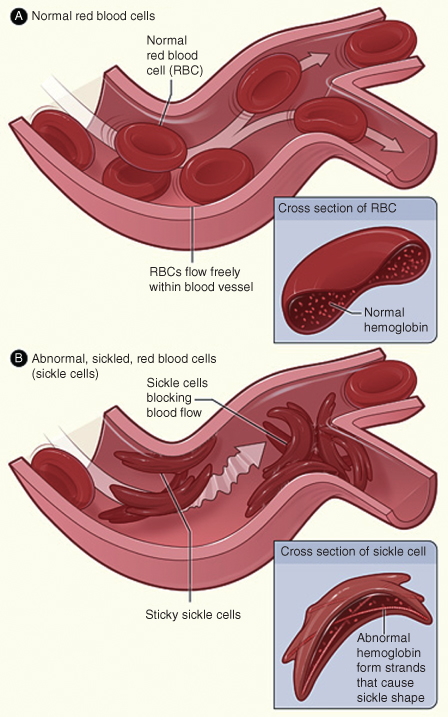
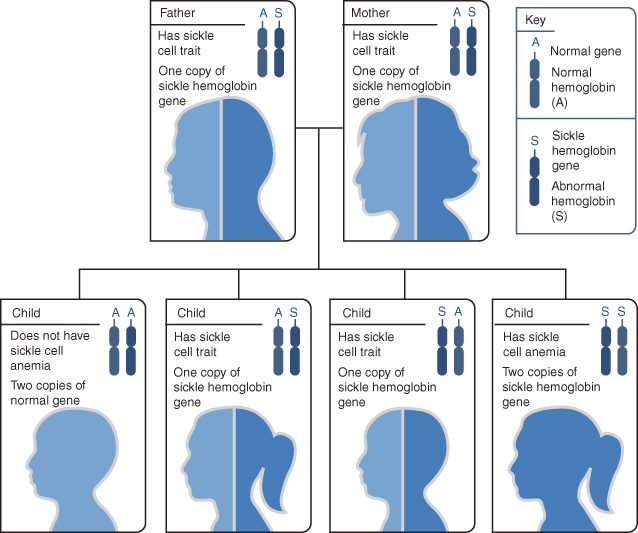
Figure 8.2 Example of an inheritance pattern for sickle cell trait. The image shows how sickle hemoglobin genes are inherited. A person inherits two hemoglobin genes—one from each parent. A normal gene will make normal hemoglobin (A). A sickle hemoglobin gene will make abnormal hemoglobin (S). When both parents have a normal gene and an abnormal gene, each child has a 25% chance of inheriting two normal genes; a 50% chance of inheriting one normal gene and one abnormal gene; and a 25% chance of inheriting two abnormal genes.
Source: National Heart, Lung, and Blood Institute; National Institutes of Health; U.S. Department of Health and Human Services. Available at: http://www.nhlbi.nih.gov/health/health-topics/topics/sca/causes.html.
Lymphoma
- Hodgkin’s Lymphoma: Overproduction and aggregation of B lymphocytes usually occurs in mediastinal, cervical, and inguinal lymph nodes, although other nodes may be affected. While the exact cause of Hodgkin’s disease is unknown, a strong association with the Epstein–Barr virus (EBV) has been reported. Other risk factors include occurrence of the disease among family members and individuals with acquired immune deficiency syndrome (AIDS).
- NHL: At the genetic level, malignant proliferation of B and T lymphocytes has been attributed to chromosomal translocations within immunoglobulin and T-cell receptor loci within these cells, respectively. The exact etiology of NHL remains unknown; however, genetic factors and environmental exposure to radiation, chemicals, herbicides, and chemotherapy have been implicated as potential causes. Helicobacter pylori, EBV, Kaposi’s sarcoma herpesvirus, and retroviruses are several infectious agents reported with increased incidence of lymphoma. Patients with autoimmune disease, posttransplant status, and human immunodeficiency virus (HIV) are also at an increased risk of developing NHLs.12
MM
- While the exact etiology of MM remains speculative, radiation and chemical exposure, and pesticides have been hypothesized as potential causes of proliferation of plasma cells and the accumulation of abnormal immunoglobulin proteins in the bone marrow.
Epidemiology
Leukemia
The epidemiology of leukemias is shown in Table 8.1. Most cases of leukemia occur in older adults with the predominant types being CML and AML. The 5-year survival rate of leukemia has increased to 55% over the time frame of 1999–2006. More males than females are diagnosed with leukemia. Leukemia causes one-third of all cancer deaths in children under age 15 years.
Table 8.1. Epidemiology of Leukemia in the United States (2010)
Adapted from The Leukemia and Lymphoma Society Facts 2010–2011.
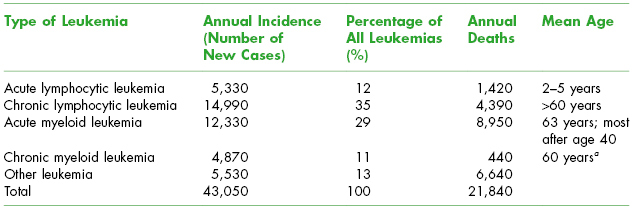
a Prognosis is less favorable when it occurs at younger ages.
Anemia
- Iron-Deficiency Anemia: Annual incidence in the United States is 5–11% in women and 2–5% in men.11 Higher incidence abounds in developing countries. Men usually do not tend to lose iron in physiological states; hence, even mild anemia in men warrants prompt evaluation.
- G6PD Anemia: The X-linked G6PD deficiency is the world’s most common enzyme disorder afflicting about 400 million people globally, especially those of Mediterranean, Middle Eastern, and Asian descent.11 This deficiency is thought to impart some degree of resistance to malaria.
- SCA: Eight to ten percent of African-Americans carry the sickle cell trait and up to 0.15% have SCA.11 Sickle cell trait imparts some degree of resistance to malaria and is endemic in several parts of Africa.
- Thalassemia: Thalassemia is more prevalent among population groups of the Middle East, Mediterranean and South Asia. The high prevalence of thalassemia among these groups has been postulated to be related to some degree of protection against malaria similar to the sickle cell trait.
- Vitamin B12– and Folate-Deficiency Anemia: The incidence of folate- and B12-deficiency-related anemias is not as high in developed countries as in other parts of the world. Folate- and B12-deficiency anemias are common in older individuals with about 2% of the population over age 60 exhibiting some form of undiagnosed megaloblastic anemia.
- AA: AA is rare and has been reported to occur with an incidence rate of about 2 per million population in the United States and the Western world.
Lymphoma
- Hodgkin’s Lymphoma: It is seen in a younger patient population with peak incidence in early adulthood, age 25–35 years. Men are affected more than women in a 3 : 2 ratio. An estimated 9000 Americans per year are diagnosed with Hodgkin’s lymphoma.9
- NHL: It comprises over 85% of lymphomas. Around 66,000 new cases of NHL are diagnosed in the United States each year and almost 20,000 deaths are attributed to it. It is a disease of adults over 65 years old.9
MM
- About 15,000 new cases of MM are diagnosed each year in the United States with a median age of 65 years and a slightly higher ratio of incidence in males.9
 Coordination of Care between Dentist and Physician
Coordination of Care between Dentist and Physician
When a dental professional suspects that a patient has a hematological disease based on history, examination, or laboratory findings, prompt referral to the primary physician is warranted for diagnosis and management of the underlying disease.
In all hematological malignancies, a dentist may be called upon by the primary team to eliminate sources of odontogenic infection prior to chemotherapy or radiotherapy and to manage oral complications associated with the disease and treatment. Dental clearance prior to HSCT is essential because infection can be life threatening in patients with severe neutropenia. Additional coordination between the dentist and oncologist is required to ensure patients in remission are medically stable to receive routine dental care with respect to their hematological status.
Oral manifestations of MM in the maxilla, mandible, and oral soft tissues are managed by the oncologist with chemotherapy and localized radiation if indicated.
 II. Medical Management
II. Medical Management
Identification/Medical History/Physical Examination
Leukemia
Medical considerations for patients with leukemias are shown in Table 8.2.
Table 8.2. Medical Considerations for Patients with Leukemia
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


