CHAPTER 21 Nonopioid Analgesics, Nonsteroidal Anti-inflammatory Drugs, and Antirheumatic and Antigout Drugs
CAUSE OF INFLAMMATION
Although inflammation is often thought of as only a pathologic event, it serves a normal homeostatic function. In the case of tissue injury from minor trauma or a dental surgical procedure, the inflammatory process results in a series of well-regulated humoral and cellular events leading to localization of injury, removal of noxious agents, repair of physical damage, and restitution of function in the injured tissue. In patients unable to mount a competent inflammatory response, such as patients with agranulocytosis induced by some cancer chemotherapeutic drug regimens (see Chapter 42), the results are often fulminant infection and death.
Inflammation can be divided into three phases: acute inflammation, subacute inflammation, and chronic inflammation. In acute inflammation, small preformed inflammatory mediators such as histamine are released, causing vasodilation and increased capillary permeability. In the subacute stage, inflammatory cells migrate and invade the site. PGs, leukotrienes, platelet-activating factor (PAF), and cytokines also play major roles. The third, or chronic, stage of inflammation involves the lymphocytic phase of injury cleansing and repair. Cytokines, especially interleukins and tumor necrosis factor-α (TNF-α), are prominent in this stage. In reality these phases are not distinct entities. Components of the subacute phase participate in the acute inflammatory process, and acute inflammatory mediators are present in chronic inflammatory disorders such as arthritis. Even so, this classification still offers a useful way to categorize this highly complex process. The following section briefly reviews some of the key mediators of the inflammatory process. Table 21-1 provides a more complete list of inflammatory mediators.
TABLE 21-1 Classification of Some Endogenous Mediators of Inflammation
| MAJOR GROUPS | MAJOR MEDIATORS |
|---|---|
| Tissue | |
| Lymphocyte products | MCP-1 |
| GM-CSF | |
| Other chemoactive factors | |
| Interferon-γ | |
| Interleukins | |
| Skin reactive factor | |
| Macrophage products | Interleukin-1 |
| Interferon-γ | |
| TNF-α | |
| PAF | |
| Mast cell products | Histamine |
| Cytokines | |
| TNF-α | |
| Leukotrienes | |
| Prostaglandin D2 | |
| PAF | |
| Eosinophil products | Lysosomal enzymes |
| Major basic protein | |
| Other cationic proteins | |
| Leukotrienes | |
| PAF | |
| Others | Reactive metabolites of oxygen |
| Endogenous pyrogens from leukocytes | |
| Leukocytosis factors | |
| Plasma | |
| Kinin system | Bradykinin |
| Complement system | C3 fragments |
| C5 fragments | |
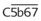 complex complex |
|
| Membrane attack complex | |
| Clotting system | Fibrinopeptides |
| Fibrin degradation products |
GM-CSF, Granulocyte-macrophage colony-stimulating factor; MCP-1, monocyte chemoattractive protein-1; PAF, platelet-activating factor.
Tissue Mediators
Histamine
Histamine is the first mediator for which a role in the inflammatory process was clearly established. This vasoactive amine is formed by decarboxylation of histidine and is widely distributed in the body. Although some free histamine exists in tissues, most is stored in mast cell and basophil granules in a physiologically inactive form. (For a more complete discussion, see Chapter 22.) Various physical and chemical stimuli, such as antigens, complement fragments, or simple mechanical trauma, can cause extrusion of the granules and release of active histamine into the extracellular fluid.
One of the most characteristic actions of histamine is dilation of vessels of the microcirculation and a marked, but transient, increase in the permeability of capillaries and postcapillary venules, reflecting an activation of H1 receptors in these tissues. These vascular changes are similar to the changes that occur in tissue after injuries of all sorts. The evidence that histamine released from the ubiquitous mast cell is responsible for the initial permeability changes seen in an inflammatory response is extensive. The histamine content of tissue fluid at the site of injury increases within minutes after the insult and then decreases. Concurrent with these changes, mast cells in the area of damage are found to be degranulated. It has been shown that prior depletion of tissue histamine stores by various means or pretreatment with classic antihistamines (H1 receptor blockers) reduces the initial vascular response to injury.101 Inhibition of initial histamine-dependent events does not block the further development of the inflammatory response, however.
In patients with chronic inflammation caused by rheumatoid arthritis, increased concentrations and penetration depth of mast cells in synovial tissue are found, possibly related to the expression of stem cell factor by synovial fibroblasts. Astemizole, a potent antihistamine, does not affect the clinical course of this disease.139 The role played by histamine in inflammation is early, transient, and nonessential for subsequent events that may lead to lasting tissue alterations.
Antihistamines have little use as general anti-inflammatory agents. In certain situations, such as immediate allergic reactions, large amounts of histamine are released locally or systemically from sensitized mast cells and basophils as a consequence of antigen-antibody reactions. In these instances, antihistamines that block the H1 receptor are useful in reducing symptoms attributable to histamine. As discussed in Chapter 22, antihistamines that block the action of histamine at the H2 receptor have a supporting role in the management of anaphylaxis and a major role in the treatment of gastric hyperacidity conditions.
Prostaglandins
The PGs are a unique family of closely related acidic lipids found in all tissues. The basic structure of all PGs is a prostanoic acid skeleton composed of a 20-carbon polyunsaturated fatty acid with a five-member ring at C8 through C12 (Figure 21-1). Similar to the leukotrienes described subsequently, PGs are derived from arachidonic acid and similar 20-carbon polyunsaturated fatty acids that are liberated from the cell membranes of local tissues by the action of acylhydrolases, principally phospholipase A2, and, in platelets, diacylglycerol lipase. Inflammatory cells, including human monocytes and mast cells, also have the ability to generate PGs. Although multiple oxygenation products are derived from arachidonic acid, the PG molecules all contain a five-membered carbon ring. The alphabetical PG nomenclature (A-F) is based on the structure of this cyclopentane ring. PGE and PGFα differ only in the presence of a ketone or hydroxyl group at C9. Also in the nomenclature, a subscript 1 indicates the presence of a double bond at C13 to C14 (PGE1), a subscript 2 indicates the presence of an additional double bond at C5 to C6 (PGE2), and a subscript 3 indicates the presence of a third double bond at C17 to C18 (PGE3).
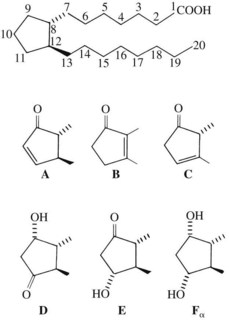
A key event in the acute inflammatory process is the liberation of arachidonic acid from damaged cell membranes on exposure to phospholipase A2 (Figure 21-2).55 This step can be inhibited indirectly by a powerful group of anti-inflammatory agents known as glucocorticoids, which are described in detail in Chapter 35. From this point, the oxidative metabolism of arachidonic acid can proceed along two divergent pathways. One pathway uses the enzyme cyclooxygenase (COX), also known as PG endoperoxide synthase, and the second pathway uses the enzyme lipoxygenase. Virtually all cells in the body, with the exception of red blood cells, contain the COX enzyme, whereas lipoxygenase seems to be limited to inflammatory cells (neutrophils, mast cells, eosinophils, and macrophages).
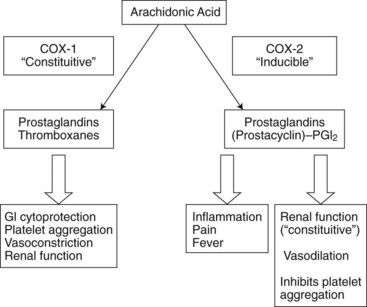
It is now well established that COX exists in two isoforms. Both are 72-kDa proteins but differ in terms of their sequence homology (approximately 60%) and their genomic regulation.47,149,150 COX-1 is regarded as the constitutive or housekeeping isoform and is the major isoform found in healthy tissues. It is always present in a number of tissues, including the central nervous system (CNS), gastric mucosa, platelets, and kidneys.36 In the gastric mucosa, COX-1 plays a major role in the synthesis of PGs involved in the formation of the mucous protective barrier (so-called cytoprotection) against stomach acid. In platelets, COX-1 is the key enzyme involved in thromboxane production and its subsequent platelet aggregatory properties necessary for proper hemostasis.
COX-2 is, for the most part, an inducible isoform upregulated by such products as cytokines, growth factors, and mitogens in human monocytes, macrophages, endothelial cells, chondrocytes, synoviocytes, and osteoblasts.36 It is associated with elevated concentrations of PGs during inflammation, pain, and fever. Figure 21-3 illustrates some of the physiologic roles of the COX isoforms. Although initially it was hoped the COX-2 products participated only in inflammatory and other pathologic processes, it is now known that oxidation products of the COX-2 isoform help regulate some normal physiologic processes, including the maintenance of adequate water and Na+ excretion by the kidneys, the inhibition of excessive platelet aggregation, and dilation of certain vascular beds.77
There is abundant evidence that PGs and the intermediate endoperoxides are mediators of inflammation.42,107 Arachidonic acid and other fatty acid precursors of PGs present in the membrane phospholipid of cells can be released by hydrolase enzymes activated by direct cellular damage or by any nondestructive perturbation of the membrane, whether physical, chemical, hormonal, or neurohumoral. PG synthesis can ensue as shown in Figure 21-2. In acute inflammatory reactions, PGs appear in fluids and exudates later (2 to 12 hours after injury) than some other mediators, such as histamine and bradykinin. PGs are being formed at a time when tissue damage and disintegration are more prominent. It is possible that some of the PG content found in sites of inflammation is derived from infiltrating neutrophils and macrophages because these cells are capable of PG synthesis.1
When released in tissue, PGs may contribute to the inflammatory response in multiple ways. The evidence that they do so can be summarized as follows42: (1) PGs are found in experimentally or naturally produced inflammatory fluids; (2) neutrophils and macrophages produce PGs during phagocytosis; (3) PGI2 and PGE2 injected intradermally are potent inducers of vasodilation and of increased vascular permeability, an effect that is greatly augmented by the presence of histamine or 5-hydroxytryptamine and may last for several hours; (4) minute amounts of PGI2 and PGE2 injected intradermally markedly increase the pain sensitivity to other mediators, such as bradykinin and histamine; (5) PGE2 is pyrogenic when injected into the cerebral ventricles or anterior hypothalamus, suggesting a mediator function; (6) a severe disabling arthritis is produced in animals by injecting PGs into the knee joint, and rheumatoid synovial cells produce PGs in culture; and (7) certain anti-inflammatory drugs that are potent inhibitors of PG synthesis reduce experimentally produced inflammation.
Leukotrienes
The term slow-reacting substance (SRS) was first applied to a lipid-soluble material produced by treatment of lung tissue with cobra venom. This material was characterized by its production of a slow, prolonged contraction of a smooth muscle preparation in contrast to the rapid and transient action of histamine. A chemically and biologically similar material was subsequently found in the lungs of sensitized guinea pigs challenged with specific antigen in vitro.153 This material was designated as the SRS of anaphylaxis (SRS-A) to distinguish it from SRS produced by nonimmunologic mechanisms. Studies of the biologic properties of SRS-A indicated that it might be an important mediator of anaphylactic and other immediate allergic reactions. SRS-A can be found in most tissues, especially in the lung, after appropriate antigenic challenge. It is released along with histamine and other active products from mast cells.
Although SRS-A was known for some time to be an acidic, sulfur-containing lipid of low molecular weight, elucidation of its exact structure and biosynthesis was delayed until intensive study of the metabolism of arachidonic acid showed that SRS-A belonged to a class of compounds known as leukotrienes.103,132 Leukotrienes are formed by the conversion of arachidonic acid to noncyclized, 20-carbon carboxylic acids with one or two oxygen substitutes and three conjugated double bonds (see Figure 21-2). The critical step in the biosynthetic pathway is generation of a 5,6-epoxide of arachidonic acid (leukotriene A4) by the combined actions of 5-lipoxygenase and leukotriene A synthase. Leukotriene A4 may be converted to leukotriene B4 by a hydrolase enzyme or, alternatively, to leukotriene C4 by the addition of glutathione. Removal of glutamate from leukotriene C4 generates leukotriene D4.
Leukotrienes C4 and D4 are potent in vivo and in vitro constrictors of bronchial smooth muscle in the guinea pig. Both compounds have similar effects in human bronchial muscle preparations, in which they are approximately 1000 times more potent than histamine. Because these leukotrienes also increase vascular permeability, it seems likely that either one or both play a role in the bronchial constriction and mucosal edema of asthma. Leukotriene B4 can enhance chemotactic and chemokinetic responses in human neutrophils, monocytes, and eosinophils.60 These findings suggest that leukotrienes may be involved in localized inflammatory processes and in asthma. Drugs that block leukotriene receptors or inhibit leukotriene synthesis by blocking the enzyme lipoxygenase are used in the treatment of asthma (see Chapter 32).
Lysosomal products
The lysosomes of neutrophils contain various enzymatic and nonenzymatic factors that play important roles in the manifestations and sequelae of inflammatory reactions (Box 21-1). During phagocytosis of bacteria or foreign material by neutrophils, the contents of lysosomes are released into the extracellular environment. They are also released on lysis of the cell. Cationic proteins from lysosomes contribute to the inflammatory process by triggering mast cell degranulation, which leads to increased vascular permeability. Other lysosomal enzymes may contribute to the inflammatory response in several ways. Several of these enzymes have the potential to damage host tissues. Collagen, elastin, mucopolysaccharides, basement membrane, and other structural elements may be degraded. Lysosomal proteases cause the production of kinin-like substances from plasma kininogen and can generate chemotactic factors for neutrophils from complement, as described in a later section. Leukotrienes and PAF are also released by neutrophils. Neutrophils may play a central role in perpetuating the inflammatory response by their dual ability to cause tissue damage and to elaborate specific mediators of inflammation. Another source of lysosomal factors, especially in chronic inflammatory lesions, may be the mononuclear phagocyte, or macrophage, the lysosomes of which contain substances similar to those of the neutrophil.1 These substances are released into the extracellular environment on activation of macrophages by various soluble factors, such as leukotriene B4, C5a complement factor, and PAF, and during the process of phagocytosis of bacteria or other particulate matter.
BOX 21-1 Factors in the Neutrophil with Inflammatory Potential
PAF, Platelet-activating factor.
| Tissue-damaging enzymes | Elastase |
| Cathepsins B and G | |
| Collagenase | |
| Other proteases | |
| Microbicidal enzymes | Myeloperoxidase |
| Lysozyme | |
| Permeability factors | Leukotrienes |
| PAF | |
| Leukokinin-forming enzyme | |
| Basic peptides | |
| Leukotactic factors | Leukotrienes |
| PAF | |
| C5-cleaving enzyme | |
| Basic peptides (chemotactic for monocytes) |
Plasma Mediators
Kinins
Bradykinin has striking pharmacologic effects in humans and animals. It is a potent but transient vasodilator of arteries and veins by a direct action on smooth muscle. Intradermal injection of bradykinin causes marked increases in vascular permeability; in this regard, it is more potent than histamine on a molar basis. Bradykinin applied to a blister base or injected intradermally or intra-arterially in humans evokes sharp pain. Experimental pain can also be produced by a bradykinin-like substance isolated from human blister fluid and from synovial fluid obtained from acutely inflamed joints. In extraction sockets, local concentrations of immunoreactive bradykinin and PGs increase in patients after the removal of impacted third molar teeth.129,159 All these phenomena implicate bradykinin in various aspects of acute inflammatory reactions, including acute pain.
Complement system
The complement system plays an important role in the inflammatory process. In humans, this system consists of at least 20 component proteins that react in a fixed sequence (Figure 21-4). An immune complex on a cell surface activates the first component, C1, and a cascade of events results in the formation of a complex that leads to membrane damage and cell lysis.
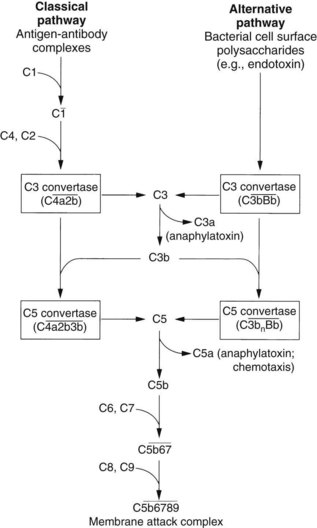
 ). The steps by which bacterial polysaccharides interact with several plasma proteins (B, D, and properdin) to generate the C3 convertase of the alternative pathway,
). The steps by which bacterial polysaccharides interact with several plasma proteins (B, D, and properdin) to generate the C3 convertase of the alternative pathway,  , are not shown.
, are not shown.Nitric oxide
The small gaseous molecule nitric oxide (NO) apparently plays a regulatory and a proinflammatory role in various inflammatory conditions, including arthritis, asthma, and inflammatory bowel disease.38,95,127 In mammalian cells (similar to COX), two isoforms of the NO-producing enzyme nitric oxide synthetase are constitutively expressed, and a third inducible isoform, inducible nitric oxide synthetase (iNOS), is upregulated in response to bacterial products and proinflammatory cytokines.92,94 Compounds that inhibit iNOS expression or activity possess anti-inflammatory properties.94,133 Currently approved drugs that target the nitric oxide system, such as nitroglycerin to treat angina (see Chapter 26) and the male erectile dysfunction drug sildenafil (Viagra) (see Chapter 28), increase NO levels; specific iNOS inhibitors are being developed to treat various inflammatory disorders.
NONSTEROIDAL ANTI-INFLAMMATORY DRUGS
NSAIDs include some of the most frequently taken medications. Because these agents share a common mechanism of action, they exert qualitatively similar therapeutic and toxic effects. For the treatment of pain and inflammation that accompany various dental surgical procedures, the short-term use (typically ≤1 week) of NSAIDs has generally proved to be highly efficacious and safe. Compared with opioid-combination drugs (described later in this chapter), NSAIDs lack various undesirable CNS depressant effects that contribute to the high incidence of drowsiness, dizziness, and nausea commonly seen with opioid-containing agents. This favorable efficacy and safety profile has led the U.S. Food and Drug Administration (FDA) to approve three NSAIDs (ibuprofen, naproxen sodium, and ketoprofen) in addition to aspirin for over-the-counter (OTC) use. Use of these drugs under OTC package insert guidelines mandates no more than 10 days of consecutive dosing for pain and only 3 days for fever, plus absolute maxima on single and daily doses that are lower than the prescription use of these drugs.81
The development of NSAIDs that are highly selective COX-2 inhibitors seemed to offer a safety advantage regarding some of the more serious adverse effects seen with long-term NSAID therapy, specifically GI ulcers, perforations, and bleeds.16,143 More recent studies showing increased cardiovascular risk with long-term use of these agents compared with placebo in colon polyp prevention trials19,151 and increased cardiovascular risk with 10 days of dosing in the treatment of patients with pain after coronary artery bypass graft (CABG) surgery119 have led to the removal of several of these drugs from the worldwide marketplace.
Salicylates
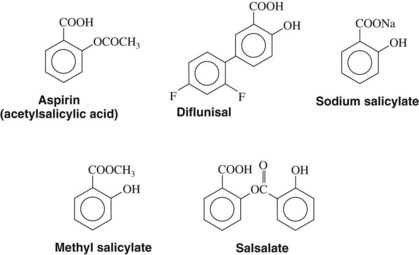
Several additional salicylates have also been marketed. Choline salicylate, magnesium salicylate, and their combination product are ion complexes of salicylate. Salsalate is an ester composed of two salicylate molecules, which are freed as the molecule is hydrolyzed. These salicylate formulations are said to have fewer GI side effects than aspirin. Because released salicylate is the active moiety of all these drugs, however, the rest of their pharmacology is quite similar to aspirin. The structure of aspirin and some of the related salicylates is shown in Figure 21-5. Aspirin may be considered a prototype of the NSAIDs and is the standard of reference against which these agents are compared and evaluated.
Mechanism of action
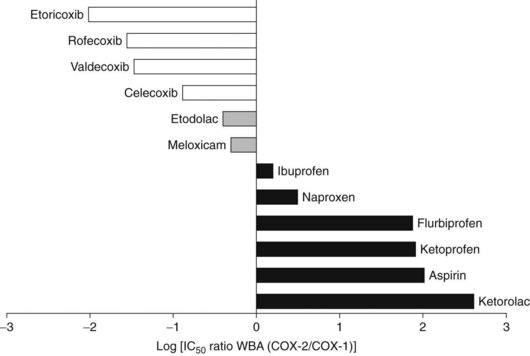
The efficacy of salicylates and all related NSAIDs as analgesic, anti-inflammatory, and antipyretic agents results from their ability to inhibit COX activity, preventing the synthesis and release of COX products, most prominently the PGs. All salicylates and almost all the currently available NSAIDs, with the exception of the highly selective COX-2 inhibitors, inhibit COX-1 and COX-2. Most of these nonselective COX-inhibiting NSAIDs, including aspirin, are more potent or at least equipotent inhibitors of COX-1,128,169 which accounts for some of the more important adverse effects of these drugs. Figure 21-6 shows the relative COX-2 versus COX-1 selectivity of some representative NSAIDs. As shown in this figure, aspirin is an approximately 100-fold more selective inhibitor of COX-1 than COX-2. The published COX selectivities of individual drugs vary with the assay system being used.
Aspirin uniquely inactivates COX by irreversibly acetylating the enzyme. Acetylation occurs on serine 530 of COX-1.105 A comparable serine on COX-2, serine 516, is also acetylated by aspirin. The later modification not only inhibits PG production during inflammation, but also enables COX-2 to produce 15-hydroxyeicosatetraenoic acid.98,123 This metabolite may be associated with certain adverse effects. Other salicylates and NSAIDs do not acetylate COX but are reversible competitive inhibitors of the enzyme. Because PGs are not stored, but rather are synthesized immediately before release, the reduction of PG concentrations by NSAIDs can be observed quickly.
The potency of the salicylates as inhibitors of PG synthesis in vitro correlates well with their ability to alleviate carrageenin-induced inflammation in animals (Table 21-2). In humans, anti-inflammatory doses of aspirin (3 g daily), sodium salicylate (3 g daily), or indomethacin (100 mg daily) reduce the output of PG metabolites in the urine by more than 75%, indicating a close correlation of the inhibition of COX with anti-inflammatory effects.71 Although inhibition by COX is the major mechanism of action of the NSAIDs, participation in other anti-inflammatory mechanisms may occur and account for some of the variation in clinical response seen with these drugs. Salicylates may inhibit cell migration and some functions of neutrophils. A reduced production of rheumatoid factor may also occur by stimulation of suppressor T-cell activity. Other mechanisms contributing to anti-inflammatory effects include reduced capillary permeability, reduced antibody production, and alterations in connective tissue synthesis. The relative potencies against COX-1 and COX-2 also account for important differences among various NSAIDs.
TABLE 21-2 Anti-inflammatory and Cyclooxygenase Inhibitory Activity of Some NSAIDs Compared with Acetaminophen

Inhibition of PG synthesis at the site of injury or inflammation can explain at least some of the analgesic effect of aspirin. Although PGs themselves do not seem to cause pain when injected locally, PGE2 and PGF2α do sensitize pain receptors to other mediators such as histamine and bradykinin. In this connection, aspirin and related drugs can prevent the writhing response elicited by bradykinin but not that produced by PGs. This finding is explained by the fact that the salicylates and all other NSAIDs inhibit the synthesis of PGs induced by bradykinin but not the binding of PGs to their receptors. Animal experiments have revealed that NSAIDs also have central analgesic actions, which may involve the inhibition of COX or other unknown mechanisms at the level of the spinal dorsal horn or at higher levels of the CNS.106,110 The antipyretic effect of aspirin and similar drugs is also mediated by the reduction of PGE2 synthesis as a result of inhibition of COX.
General therapeutic effects
Acute pain
It is difficult to separate the analgesic and anti-inflammatory effects of NSAIDs because most painful conditions have an inflammatory component. There is little doubt that the cascade of reactions leading to the formation of PGs is integrally involved with the inflammatory response and that aspirin’s efficacy in treating inflammation and pain is closely related to its inhibition of PG synthesis. By using microdialysis techniques, it has been shown that after dental surgery the analgesic effects of NSAIDs correlate with a local reduction in PG synthesis.121,129 Nevertheless, several observations suggest that the analgesic and anti-inflammatory effects of NSAIDs may occur by different mechanisms. First, there is a different time course for the onset of analgesic and anti-inflammatory effects. Clinically significant analgesia usually occurs within 1 hour of drug administration, whereas anti-inflammatory effects sometimes take several days or weeks to reach maximum levels because chronic inflammatory processes may be occurring that cannot be quickly reversed by inhibiting PG production. Also, the maximal human analgesic effect usually occurs at lower doses than the antirheumatic and other anti-inflammatory effects.
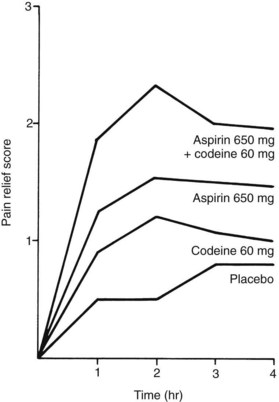
Aspirin is an effective analgesic for almost any type of acute dental pain. Double-blind, controlled studies of the relief of pain after the surgical extraction of third molars have shown that 650 mg of aspirin is substantially more effective than 60 mg of codeine in relieving postoperative pain (Figure 21-7).31 Most controlled clinical studies have established that, regardless of the cause of the pain, aspirin (650 mg) provides equal or greater pain relief than codeine in standard doses (60 mg).12 Aspirin and other NSAIDs and acetaminophen have what is known as a ceiling or plateau effect in the treatment of acute pain. In other words, there is a dose-response for pain relief up to 650 to 1000 mg of aspirin, but increasing the dose beyond these amounts does not enhance the analgesic effect further and does increase the likelihood for toxic effects.
Rheumatic fever
One of the early uses of salicylates was in the treatment of rheumatic fever. Aspirin markedly reduces the acute inflammatory components of the disease, such as fever, joint pain, swelling, and immobility. Salicylates do not affect other aspects of the disease, however, such as the proliferative reaction in the myocardium leading to scarring, and they do not alter the progression of the disease. Although anti-inflammatory drugs, including corticosteroids (see Chapter 35), may be used to reduce inflammation, antibiotic therapy is the major therapeutic strategy.
Rheumatoid arthritis
Rheumatoid arthritis is a chronic systemic disease of unknown origin. Several tissues and organs may be involved, but in most patients the chief clinical and pathologic features result from chronic inflammation of synovial membranes. Irreversible joint injury (subluxation, loss of motion, or ankylosis) results from formation of chronic granulation tissue that causes erosions of articular cartilage, subchondral bone, ligaments, and tendons. Extra-articular manifestations, such as subcutaneous or subperiosteal nodules of granulation tissue, peripheral neuropathy, and chronic skin ulcers, occur to a variable extent and seem to result from generalized focal vasculitis. Patients with rheumatoid arthritis are at increased risk of developing cardiovascular disease, including myocardial infarction (MI).168
Salicylates (usually as aspirin) are still widely used in the clinical management of rheumatoid arthritis. The pain and inflammation of rheumatoid arthritis can often be controlled with salicylates alone (or with another NSAID).152 Salicylates produce a measurable reduction of inflammation in the joints and associated tissues, a lessening of symptoms, and improved mobility. Salicylates may also reduce neutrophil activity in addition to inhibiting PG synthesis. Clinical observation suggests that salicylate therapy can diminish or delay the development of crippling associated with the disease. This benefit is not a direct effect of the drug on the progression of the disease, but relates more to reduction of pain and subsequent facilitation of mobility. In addition to salicylates, the basic therapeutic regimen in rheumatoid arthritis includes rest, physical measures (primarily heat), and exercise.
Fever
Fever is typically a symptom of a disease process, usually a viral or bacterial infection brought about by exogenous (microbial products) and endogenous pyrogens.40,156 It is believed that exogenous pyrogens stimulate host cells to produce endogenous pyrogens, of which IL-1 has been the best characterized.51,156 IL-1 is thought to act on the anterior hypothalamus, generating the local release of PGs.51,155 Injection of PGs into the brain of various animal species is known to increase body temperature.53 The PGs and possibly other non-PG endogenous pyrogens elevate the thermal set-point of the body so that it retains the ability to regulate closely its temperature, but at a higher temperature than normal.40,156
Prophylaxis against platelet aggregation
The correlation between the ability of aspirin-like compounds to inhibit simultaneously platelet aggregation and the production of PGs has been known for almost 40 years.148 Aspirin inhibits the synthesis of TXA2 by irreversible acetylation of the COX enzyme in platelets.73,130 Lacking a nucleus, platelets cannot generate new enzyme, and the ability of affected platelets to produce TXA2 is permanently blunted during their 10-day lifetimes. Most platelet COX acetylation may occur presystemically as platelets pass through gut capillaries before the hydrolysis of aspirin to salicylate (a weak and reversible inhibitor of COX) in the gut and the liver.125 This possibility may partially explain the relative lack of effect of low-dose aspirin on the antiaggregatory PGI2 molecule produced by the systemic vascular endothelium.
In 1998, the FDA published a set of indications for aspirin prophylaxis that include definitive dosing guidelines for each condition (Table 21-3).44 Vascular indications include the reduction of death or nonfatal stroke in patients who have had ischemic strokes or transient ischemic attacks, reduction of vascular mortality in patients with acute MI, reduction of combined risk of death and nonfatal MI in patients with previous MI or unstable angina, and reduction of the combined risk of MI and sudden death in patients with chronic stable angina. The recommended daily dose for all these indications is low (≤325 mg/day) compared with the analgesic and anti-inflammatory uses of the drug. Aspirin also is indicated for patients who have undergone revascularization procedures, including CABG surgery, percutaneous transluminal coronary angioplasty, and carotid endarterectomy, when there are preexisting conditions for which aspirin is already indicated.
TABLE 21-3 Antiplatelet Indications and Dosing Guidelines for Aspirin
| INDICATIONS | RECOMMENDED DAILY DOSE | DURATION OF THERAPY |
|---|---|---|
| Vascular Indications | ||
| Ischemic stroke and TIA | 50-325 mg | Indefinitely |
| Suspected acute MI | 160-162.5 mg taken as soon as MI is suspected, then once daily | For 30 days after MI (after 30 days, consider further treatment if previous MI) |
| Prevention of recurrent MI | 75-325 mg | Indefinitely |
| Unstable angina pectoris | 75-325 mg | Indefinitely |
| Chronic stable angina pectoris | 75-325 mg | Indefinitely |
| Revascularization Procedures in Selected Patients | ||
| CABG surgery | 325 mg starting 6 hr after procedure | 1 yr |
| PTCA | 325 mg 2 hr before surgery; maintenance therapy 160-325 mg | Indefinitely |
| Carotid endarterectomy | 80-650 mg twice daily started before surgery | Indefinitely |
CABG, Coronary artery bypass graft; MI, myocardial infarction; PTCA, percutaneous transluminal coronary angioplasty; TIA, transient ischemic attack.
Absorption, fate, and excretion
When aspirin is taken orally, it is rapidly absorbed from the stomach and small intestine. Aspirin is a weak acid, with a pKa of approximately 3.5, which favors its absorption in the stomach. Most absorption occurs in the small intestine, however, because of its much larger surface area. The rate-limiting steps in the absorption of aspirin are the disintegration and dissolution of the tablet. These two steps can be greatly influenced by the manufacturing process, which determines factors such as particle size and compression of the tablet. Buffering the tablet increases the rate of dissolution, but the fastest absorption is obtained when aspirin is dissolved in hot water before ingestion. Other factors, such as gastric emptying time, gastric contents, and level of autonomic activity of the patient, may also influence the rate of absorption.100,102
Aspirin has a 15-minute half-life.124 Because its acetylation of COX in the platelet is irreversible, however, the full extent of its antiplatelet action depends on the life span of the platelet (8 days) and not on the short half-life of aspirin.4,124 It is quickly metabolized by gastric and plasma esterases to salicylate ion (Figure 21-8). Although some aspirin becomes bound to plasma proteins, 80% to 90% of the salicylate ion is bound for a short time, principally to albumin. Salicylate is distributed throughout most body fluids and tissues. It can be isolated from spinal, peritoneal, and synovial fluids; saliva; breast milk; and sweat. Salicylate freely crosses the placenta from mother to fetus.
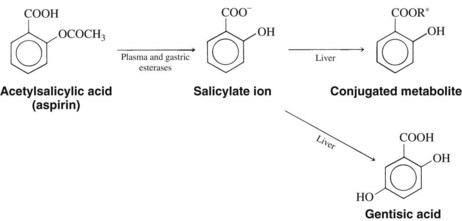
The elimination half-life of sodium salicylate is 2 to 3 hours after a single analgesic dose. The liver is the main site of biotransformation, and conjugation is the primary route. Because the metabolism of salicylate is capacity limited, large repeated doses or single toxic ingestions result in plasma half-lives of 5 to 30 hours. When describing the duration of analgesic and anti-inflammatory action of aspirin, its ultimate dosing interval reflects the summed half-lives of the parent molecule and the active metabolite, salicylate. Salicylate, in contrast to the parent aspirin molecule, is only a reversible inhibitor of COX (most notably in the platelet), however. The three primary products of salicylate conjugation are salicyluric acid (the glycine conjugate), the ether or phenolic glucuronide, and the acyl or ester glucuronide. A small portion (<1%) is oxidized to gentisic acid (see Figure 21-8). Free salicylate and the salicylate metabolites are excreted by glomerular filtration and by active proximal tubular secretion in the kidney. In normal humans, approximately 10% of ingested salicylate appears unchanged in the urine; however, this fraction may decrease to 2% or increase to 30% with urinary acidosis or alkalosis. A higher percentage of free salicylate is excreted at higher doses because the liver is unable to maintain the same percentage of metabolism at higher doses of salicylates.
Adverse effects
With more long-term, high-dose regimens of aspirin and other nonselective NSAIDs used in the treatment of various inflammatory disorders, the inhibition of COX-1 leads to several common and predictable effects, the occurrence of which varies with each drug. The inhibition of PGI2 and PGE2 synthesis and the resulting loss of their protective effects on the gastric mucosa lead to a significant increase in GI problems, the most serious of which include significant gastric bleeding, symptomatic peptic ulcers, and GI perforations and obstructions. The incidence of these more serious events ranges from 1% to 5% of patients per year. Even long-term low-dose (81 mg/day) aspirin therapy is associated with an increased risk of serious GI events, at a rate of 0.1% to 0.2% of patients per year.124 This increase in GI complications has led experts to question the widespread use of low-dose aspirin therapy in patients without significant cardiovascular or cerebrovascular risks. Nonaspirin salicylates generally elicit fewer adverse effects in the GI tract but cannot be used for platelet antiaggregatory therapy. Most of the other NSAIDs have some antiplatelet effect based on inhibition of the production of TXA2, but the effects are not as pronounced as with aspirin because aspirin is an irreversible inhibitor of COX.
The adverse effects of aspirin and other NSAIDs on the kidney are well known. Normal renal function is partly dependent on PG synthesis. It is believed that COX-1 and COX-2 are important in producing PGs involved in reducing water and Na+ reabsorption at the ascending loop of Henle and maintaining proper dilation of the renal vasculature (see Figure 21-3).17 With NSAID therapy, dose-dependent water and Na+ retention manifested by peripheral edema, elevation in blood pressure, and rarely congestive heart failure is thought to follow the inhibition of PGE2 synthesis. Renal artery vasoconstriction, possibly causing acute renal ischemia and kidney failure, is ascribed to an inhibition of PGI2 synthesis. Acute renal failure is more likely to develop in patients with preexisting renal insufficiency, congestive heart failure, or dehydration because the renal arterioles are more dependent on PGs to maintain normal perfusion of the glomeruli.117 Acute renal failure is also more likely when NSAIDs are given concurrently with an angiotensin-converting enzyme (ACE) inhibitor because the lack of angiotensin II weakens reactive constriction of the efferent arteriole, a normal protective response for maintaining glomerular filtration in patients with reduced renal blood flow.
The use of aspirin in children with viral infections has been associated with Reye’s syndrome.84 First described in 1963, Reye’s syndrome is an acute childhood illness that produces metabolic encephalopathy and liver disease.126 Typically, a child is recovering from influenza or varicella when the acute encephalopathic symptoms of lethargy, agitation, delirium, and seizures appear. Without aggressive supportive treatment, the disease progresses to deep coma, brainstem dysfunction, and death in 80% to 90% of cases.84 Even with heroic treatment, the mortality rate can exceed 30%, and survivors can be left with permanent brain damage. Clinical reviews and case-controlled studies performed in the 1970s and 1980s reported a strong association between the development of Reye’s syndrome and the ingestion of aspirin in children and teenagers with viral infections.85,104,171 Aspirin and related salicylates are contraindicated for the treatment of flulike symptoms, chickenpox, gastroenteritis, and, in the opinion of most pediatricians, any febrile respiratory condition in children or teenagers. Acetaminophen and ibuprofen have not been associated with the development of Reye’s syndrome.81
The cardinal signs and symptoms of acute aspirin overdose include nausea, vomiting, tinnitus, hyperthermia, and hyperventilation. Hyperventilation arises in part from a direct stimulation of respiratory centers in the brain and from a compensatory increase in respiration in response to excessive carbon dioxide produced by large doses of aspirin partially uncoupling oxidative phosphorylation. This uncoupling also accounts for the paradoxic (because therapeutic doses of aspirin are used for antipyresis) increase in body temperature.18 The hyperventilation eventually can lead to respiratory alkalosis, which may be followed by a combined respiratory and metabolic acidosis accompanied by dehydration. Acidosis is more prominent as the level of overdose increases. Acidosis is also more likely to occur in children and infants. Impaired vision, hallucinations, delirium, and other CNS effects may be evident, and the situation is considered life-threatening.
Intolerance to salicylates can occur, with symptoms ranging from rhinitis to severe asthma. This reaction does not seem to be immune mediated even though it resembles drug allergy in clinical presentation. Aspirin intolerance is more common in patients with preexisting asthma or nasal polyps. The incidence of this reaction in asthmatic patients has been reported as high as 20%. Patients with a history of asthma, allergic disorders, or nasal polyps should be questioned to ensure that they can tolerate aspirin and other NSAIDs. The bronchoconstriction may be caused by a shift in the arachidonic acid cascade when the COX enzyme is blocked. This inhibition prevents arachidonate metabolism from producing bronchodilating PGs, primarily PGE2.160 The lipoxygenase pathway predominates and produces leukotrienes that constrict bronchioles in sensitive individuals, mimicking the asthmatic attack.22,39 Other manifestations of aspirin intolerance include urticaria (hives) and angioedema. Switching from aspirin to another salicylate or even another NSAID does not prevent the reaction; acetaminophen is the only antipyretic analgesic that may be used safely in patients with aspirin intolerance. The clinician should be aware that, although relatively rare, there are reports of aspirin-intolerant asthmatic patients also displaying severe respiratory symptoms when ingesting therapeutic doses of acetaminophen.7
Contraindications and precautions
Aspirin is contraindicated or at least must be used with caution in numerous medical conditions (Table 21-4). Serious internal bleeding can result from the ingestion of aspirin by a patient with an ulcer. Patients with compromised liver function should use aspirin cautiously because, when used on a long-term basis, aspirin increases the prothrombin time, which could aggravate bleeding problems. Low doses of aspirin can increase plasma urate concentrations and exacerbate gouty arthritis as a result of competition between salicylate and uric acid at the active secretion sites in the proximal tubule of the kidney or by an increase in uric acid reabsorption. High doses of aspirin may increase or decrease plasma glucose concentrations by stimulating epinephrine and glucocorticoid release or by depleting liver glycogen. Salicylates may also increase insulin secretion because PGE2 inhibits insulin secretion.66
TABLE 21-4 Contraindications to the Use of Aspirin and Other Salicylates
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


